Глава 13 термокаталитические превращения углеводородов нефти и газа
Глава 13
ТЕРМОКАТАЛИТИЧЕСКИЕ ПРЕВРАЩЕНИЯ УГЛЕВОДОРОДОВ НЕФТИ И ГАЗА
•
Увеличение объема производства нефтепродуктов, расширение их ассортимента и улучшение качества в условиях, когда непрерывно возрастает доля переработки сернистых, высокосернистых и высокопарафинистых нефтей, потребовало ускоренного развития вторичных и особенно каталитических процессов. В СНГ с помощью катализаторов производят в настоящее время около 75 % всех продуктов химической, нефтеперерабатывающей и нефтехимической промышленности. Из новых химических процессов на применении катализаторов основано более 90 %. В нефтепереработке наиболее распространены каталитические процессы получения топлив — каталитический крекинг, риформинг, гидроочистка, алкилирование, изомеризация и гидрокрекинг. Каталитические процессы гидроочистки и гидрокрекинга используют также для производства высококачественных нефтяных масел и парафинов.
13.1. ОБЩИЕ СВЕДЕНИЯ О КАТАЛИЗЕ И КАТАЛИЗАТОРАХ
По характеру взаимодействия катализатора с реагирующими веществами и по типу промежуточных продуктов различают окислительно-восстановительные и кислотно-основные реакции и соответственно катализаторы.
Многие промышленные катализаторы являются бифункциональными, так как окислительно-восстановительный катализатор наносят на кислотный носитель. С другой стороны, многие сульфиды и оксиды сами по себе обладают и окислительно-восстановительной, и кислотио-основной активностью.
Наибольшее распространение в нефтеперерабатывающей промышленности получил гетерогенный катализ активной поверхностью твердого тела.
Активность, селективность и стабильность катализаторов. Одна и та же реакция может протекать в присутствии различных катализаторов. Скорость данной реакции в зависимости от природы катализаторов характеризует их активность. НапрИ7 мер, относительная константа скорости гидрирования этилена в присутствии различных катализаторов составляет:
Cr 1 Pt 100
Ni 13 Pd 1000
Rh 1800
Таким образом, наиболее активным катализатором гидрирования этилена является родий.
В подавляющем большинстве случаев в присутствии катализатора помимо основной протекает еще ряд параллельных и последовательных реакций. Доля исходных веществ, превращаемая в целевой продукт, характеризует селективность катализатора. Селективность реакции на данном катализаторе зависит также от условий процесса.
Важнейшим свойством катализатора является его способность сохранять активность во времени, характеризуемая стабильностью. При гомогенном катализе жидкий катализатор дезактивируется в процессе работы в результате накопления в нем продуктов, снижающих его концентрацию.
Значительно многообразнее причины снижения активности твердых катализаторов. Твердые катализаторы претерпевают как физические, так и химические изменения. При длительном воздействии температуры происходит рекристаллизация металлов, приводящая к изменению удельной поверхности катализа^ тора или числа активных центров. Для повышения устойчивости катализаторов к рекристаллизации в его состав вводят небольшие добавки веществ — структурообразующих промоторов, снижающих скорость рекристаллизации. Механические и термические воздействия приводят также к постепенному разрушению гранул катализатора. Химические изменения катализаторов вызваны хемосорбцией на их поверхности примесей к сырью или продуктов их разложения. Примеси, отравляющие катализатор, называются ядами. В процессах нефтеперера* ботки ими обычно являются соединения серы, азота и других гетероатомов, а также металлорганические соединения, содержащиеся в сырье. При каталитической переработке углеводородов на поверхности катализатора постепенно накапливается кокс. Отложения кокса, покрывая активную поверхность катализатора, прекращаю-]' доступ к ней молекул сырья. Удаленйё коксовых отложений с поверхности катализатора Осуществляют кислородом воздуха, диоксидом углерода или водяным паром в процессе регенерации.
Механизм действия катализаторов окислительно-восстановительного типа. Специфика каталитических реакций состоит в том, что обмен электронами между реагирующими связями осуществляется с участием электронов катализатора. Типичными катализаторами окислительно-восстановительных реакций являются переходные металлы и полупроводники.
Активность переходных металлов (Fe, Со, Ni, Ru, Rh, Re, Ir, Pt и др.) в окислительно-восстановительных реакциях объясняется незавершенностью их ^-оболочек. Неспаренный электрон незавершенной d-орбитали действует как «свободная валентность», в значительной мере подобно свободному радикалу. Если адсорбированная молекула имеет незанятые орбитали, то возникает ковалентная связь за счет перехода электрона из металла на вакантные уровни молекулы. Если поверхность металла обладает большим сродством к электрону по сравнению с адсорбированной частицей, то происходит переход электрона от молекулы в металл. Между этими крайними механизмами возможны различные переходные формы. Полагают, что образующиеся поверхностные хемосорбированные соединения имеют много аналогий с комплексными соединениями. В настоящее время на основании этого предположения пытаются объяснить повышенную реакционную способность хемосорбированных молекул с помощью теории поля лигандов и трактовать возникающие связи органических молекул с катализатором как я-связи.
Образование промежуточного соединения катализатора с молекулой, обладающей неподеленной парой электронов, может происходить путем переноса заряда от молекулы к незаполненной орбитали металла с получением так называемого комплекса с переносом заряда. Соединения этого типа можно рассматривать как молекулярные донорно-акцепторные соединения. Связь, возникающая в комплексах с переносом заряда, носит обычно полярный характер.
В случае полупроводников (оксиды, сульфиды никеля, молибдена, вольфрама и некоторых других переходных металлов) свободные валентности (свободные электроны и электронные дырки) появляются вследствие неполной координированности атомов поверхности кристаллической решетки и в результате различных дефектов кристалла полупроводника. Например, узел кристалла, в котором отсутствует катион, ведет себя как отрицательный заряд, отталкивая электроны в ближайших узлах. В результате эти электроны могут быть вытеснены из валентной зоны в зону проводимости. Появление электронов (или дырок) в зоне проводимости может быть вызвано также присутствием в кристалле различных примесей, обладающих электро-нодонорными (или электроноакцепторными) свойствами, а также нарушениями стехиометрического состава. На поверхности кристалла электроны (или дырки) проводимости будут играть роль свободной валентности или активных центров.
При взаимодействии молекулы углеводорода со свободными электронами (дырками) поверхности молекула подвергается диссоциации:
R
Н

§Н§Г^Блумсдающий '—J электрон
В результате взаимодействия одна часть молекулы связывается с катализатором прочной двухэлектронной связью, другая— слабой одноэлектронной. Вследствие непрерывно протекающих процессов электронного обмена эти связи могут взаимно переходить друг в друга. Малая стабильность и высокая реакционная способность образующихся поверхностных соединений обуславливают высокую скорость их дальнейших превращений.
Каждая молекула может реагировать с данным активным центром различными путями. В одном элементарном каталитическом акте могут принимать участие группы из двух, трех и более центров (дублеты, мультиплеты), как постулировалось в теории Баландина и как было показано для многоцентровых комплексов переходных металлов. Таким образом, каталитическая активность прямо связана с числом свободных валентностей на поверхности катализатора.
Окислительно-восстановительные катализаторы используют в процессах риформинга, гидроочистки и гидрокрекинга для увеличения скорости реакций гидрирования — дегидрирования.
Кислотный катализ. Катализ жидкими и твердыми кислотами широко применяют в нефтеперерабатывающей промышленности. Каталитическое действие кислот обусловлено образованием при их взаимодействии с углеводородами катионов, называемых карбоний-ионами или карбкаТионами. Обычно карб-катионы образуются при передаче протона от катализатора (кислота НХ) к молекуле ненасыщенного углеводорода:
НХ + СНзСН — CHR —*¦ CH.,CH2CH+R + Х-.
При достаточно высокой кислотности катализатора карбка-тионы образуются также из алканов или циклоалканов путем гетеролитического разрыва связей в молекуле под воздействием катализатора (кислота Льюиса L):
СцНая+а + L ¦ *• (CnHjn+i)+ Ч" LH".
Карбкатионы — чрезвычайно реакционноспособные соединения. Константы скорости ионных реакций на несколько порядков выше аналогичных радикальных реакций. Об относительной устойчивости карбкатионов можно судить по теплоте их образования (в кДж/моль):
|
СНз | 1092 |
СН3СН2СНСН3 | 765 |
| 4- | 4- | ||
| СНзСН2 | 916 | (СНз)зС | 706 |
| -+¦ | + |
||
| СНзСН2СН2 | 869 | С.Н* |
1105 |
| CH3CH2CH2CH2 |
844 | С6Н5СН2 | 897 |
Стабильность карбкатионов возрастает в последовательности: первичный < вторичный < третичный.
Основными реакциями карбкатионов, как и радикалов, являются мономолекулярный распад по p-правилу и бимолекулярные реакции замещения и присоединения. Существенное отличие карбкатионов от радикалов — их способность к изомеризации, что объясняется значительным снижением запаса свободной энергии при переходе от первичного к вторичному и третичному карбкатионам.
Из кислотных катализаторов наибольшее распространение в нефтеперерабатывающей и нефтехимической промышленности получили алюмосиликаты, галогениды алюминия, бора, сурьмы, оксид алюминия, сульфиды некоторых переходных металлов, а также ряд протонных кислот. Кислотные катализаторы используют в процессах каталитического крекинга, риформинга, изомеризации и других для ускорения реакций, протекающих по карбкатионному механизму.
Реакции карбкатионов. Изомеризация. Изомеризация карбкатионов может происходить в результате переноса как гидрид-иона, так и метиланиона:
| СН2СНСН2СН3 СНэСНСНгСНз + 79 кДж/моль.
4--СН з
СН3СНСН2 :СЙ3| СНз^НСН*
СНз
СН3ССН3 + 59 кДж/моль.
+
Распад по р-связи. Расщепление карбкатионов обычно происходит по наиболее слабой p-связи С—С. Реакция эндотер-
мична:
-—v СН2 = СН2 -+- СН2СН2СН2СН2СН2СН3 — 92 кДж/моль.
Склонность к распаду-снижается при переходе от первичного иона ко вторичному и от вторичного к третичному. Если для распада первичного октйлкатиона требуется 92 кДж/моль, то для вторичного октилкатиона надо затратить 176 кДж/моль.
Склонность к распаду возрастает, если в результате реакции образуется не первичный, а вторичный или третичный ион:
СН3СН2СНСН2С (СНзЬ СН2СН3 —>
—* СНзСН2СН = СН2 + С(СНз)2СН2СНз- 21 кДж/моль.
Сопоставление энергетики распада и изомеризации карбка-тионов показывает, что изомеризация должна предшествовать в большинстве случаев распаду. Преимущественное образование третичных карбкатионов и их устойчивость приводят к накоплению изоструктур при каталитическом крекинге алканов.
Присоединение карбкатионов к алкенам и аренам. Эта реакция обратна реакции распада карбкатио: нов:
(СН3)зС+ СН2 = С (СНз)2 —> (СНз)зССН2С (СН3)2.
Поэтому характер изменения теплового эффекта противоположен реакции распада.
Передача протона молекуле алкена или аниону катализатора. Например:
СН2СН (СН3) СНз + СНзСН СНСНз — >
—^ СН2 = С(СНз)СНз + СНзСНСН2СН3+ 73 кДж/моль.
Наиболее энергетически выгодна такая реакция, когда протон отщепляется от первичного карбкатиона, а в результате Образуется третичный карбкатион. При передаче протона катализатору происходит обрыв цепи.
Отрыв гидрид-иона от молекулы углеводорода. Например:
СНзСНСН2СНз + СНзСН (СНз) СНз —>
—> СНзСН2СН2СН;! + СНзС (СНз) СНз + 41 кДж/моль.
Таким путем обычно осуществляется передача, цеци.
Активность карбкатиона в реакции отрыва гидрид-иона от молекулы углеводорода также снижается в ряду:
^ Р^“ R+
кперв Аетор 1Хтрет‘
Карбкэтионные реакции всегда протекают или в. жидкой фазе, или на поверхности твердого катализатора. Сольватация в растворе и адсорбция при реакции на твердой поверхности значительно изменяют тепловые эффекты реакций ионов. В результате соотношения тепловых эффектов реакций разных карбкатионов в реальных процессах могут существенно отличаться от расчетных соотношений в газовой фазе.
13.2. КАТАЛИТИЧЕСКИЙ КРЕКИНГ
Каталитический крекинг —процесс каталитического деструктивного превращения тяжелых дистиллятных нефтяных фракций в моторные топлива и сырье для нефтехимии, производства технического углерода и кокса. Процесс протекает в присутствии алюмосиликатных катализаторов при температуре 450— 530 °С и давлении 0,07—0,3 МПа.
Механизм большинства реакций каталитического крекинга удовлетворительно объясняется в рамках цепной карбкатион-ной теории. В условиях каталитического крекинга карбкатионы могут существовать только в виде ионных пар карбкатион — отрицательно заряженный активный центр поверхности.
Химические основы процесса. Сущность процессов, протекающих при каталитическом крекинге, заключается в следующих реакциях:
1) расщепление высокомолекулярных углеводородов (собственно крекинг)1;
2) изомеризация;
3) дегидрирование циклоалканов в арены.
Деструкция тяжелого нефтяного сырья вызывает образование дополнительного количества светлых моторных топлив, наибольшее значение из которых имеет бензин. Реализация всех трех типов реакций приводит к повышению октанового числа бензина: при одинаковой структуре октановые числа углеводородов возрастают с уменьшением молекулярной массы; октановые числа изоалканов выше, чем алканов нормального
Таблица 13.1. Октановые числа углеводородов,
определяемые моторным и исследовательским методами
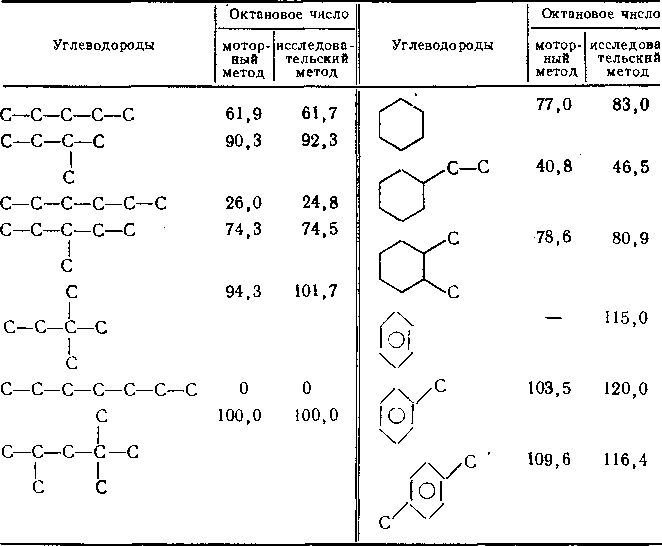
строения, а аренов — выше, чем циклоалканов :и алканов (табл. 13.1).
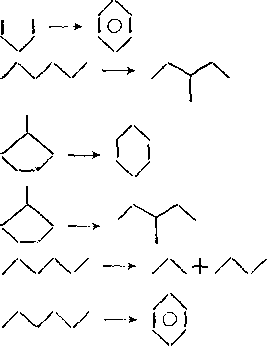
Превращения алканов. Алканы в условиях каталитического крекинга подвергаются изомеризации и распаду на алканы и алкены меньшей молекулярной массы.
Первая стадия цепного процесса — зарождение цепи — может происходить двумя способами.
При первом способе часть молекул алканов подвергается вначале термическому крекингу. Образующиеся алкены отры-' вают-протоны от катализатора и превращаются в карбкатионы:
RCH = CH2 + HA —RCHCHs + A'.
По второму способу образование карбкатиоиа возможно непосредственно из алкана путем отщепления гидрид-иона под действием протонного центра или апротонного катализатора: RH + H* —> R+ + H2; RH + L —> R+ + LH~.
Ввиду того, что отрыв гидрид-иона от третичного углеродного атома требует меньших затрат энергии, чем от вторичного и первичного, изоалканы крекируются значительно быстрее, чем алканы нормального строения.
Реакции развития цепи включают все возможные в данных условиях реакции карбкатионов. Например, если на первой
стадии процесса образовался первичный карбкатион С7Н15, то наиболее вероятным направлением его превращения будет изомеризация в более устойчивые вторичную и третичную структуры. Теплота, выделяющаяся при изомеризации, может быть затрачена на расщепление нового иона. Таким образом, про-
+
цесс превращения карбкатиона С7Н15 состоит в последовательно-параллельном чередовании реакций изомеризации и р-рас-пада:
СН3СН2СН2СН2СН2СН2СН2
|| изомеризация
СНзСН2СН2СН2СН2СНСНз
P'pacna*
СН3СН2СН2СН2 + [
СН2 = СНСНз |
л
изомеризация
+
р-распад
СНзСН2СН2СН2СНСН2СНз —--- СНзСН2СН2 + | СН2— СНСН2СН31
изомеризация
СНзСН2СН2СН2ССНз
Р~распадн-
СНзСН2СН2 + СН'з
СН2 = С —СНз СНз
Так как распад алкильных карбкатионов с образованием первичных и вторичных ионов Ct—Сз происходит значительно труднее, чем с образованием третичных ионов с большим числом атомов углерода, то скорость каталитического крекинга алканов возрастает с удлинением цепи. Например, при крекинге в одинаковых условиях степень превращения С5Н12 составляет 1 %; C7Hie — 3 %; С12Н24— 18 %; CieH34 — 42 %. Легкость (низкая эндотермичность) распада ионов с отщеплением третичных карбкатионов приводит к накоплению изоструктур в продуктах распада алканов, содержащих 7 и более атомов углерода.
Выделяющиеся низкомолекулярные карбкатионы после изомеризации отрывают гидрид-ион от молекулы исходного углеводорода, и весь цикл реакций повторяется. Обрыв цепи происходит при встрече карбкатиона с анионом катализатора:
R+ + LH- —> RH + L.
Скорость каталитического крекинга алканов на 1—2 порядка выше скорости их термического крекинга.
Превращения циклоалканов. Скорость каталитического крекинга циклоалканов близка к скорости крекинга алканов с равным числом атомов углерода. Основными реакциями циклоалканов являются: раскрытие кольца с образованием алкенов и диенов; дегидрирование, ведущее к образованию аренов; изомеризация циклов и боковых цепей.
Стадия инициирования — возникновения карбкатионов — для насыщенных углеводородов циклического и ациклического строения протекает одинаково.
Образовавшиеся карбкатионы отрывают гидрид-ион от молекул циклоалканов. Отщепление гидрид-иона от третичного углеродного атома протекает легче, чем от вторичного, следовательно, глубина крекинга возрастает с увеличением числа заместителей в кольце:
Строение молекулы
СНз СНз Н3С СНз
\/
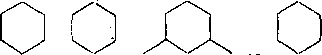
СНз
I НзС СНз
75,6
78,6 5t,8
Глубина крекинга, % 47
Неоструктуры (1,1-диметилциклогексан) отщепляют гидрид-ион от вторичного углерода, поэтому степень превращения близка к незамещенному циклогексану.
Распад циклогексильного иона может происходить двумя путями: с разрывом С—С-связей и с расщеплением С—Н-связей.
В результате реакции с разрывом С—С-связей образуются алкены и алкадиены по следующему механизму:
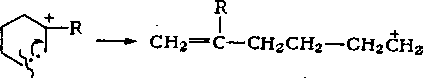
СН2 = С (R) СН2СН2СН2СН2, —* СН2 = С (R) СНСН2СН2СН3.
+
+
Алкенильный ион легко изомеризуется в аллильный:
Наиболее вероятными реакциями аллильного иона являются отрыв гидрид-иона от исходной молекулы
+
СН2 = С (R) СНСНаСНаСНз + 'RH - >
+
СН2 = С (R) СНСН2СН2СНз + "RCH = СН;
+
I СН2 = С (R) СН= СН - СН2СНз | + "RCHCH3.
Распад циклогексильного карбкатиона с расщеплением С—Н-связей энергетически более выгоден, так как через про-, межуточные циклоалкеновые структуры образуются арейы:
Циклоалкены подвергаются каталитическому крекингу значительно быстрее, чем циклоалканы:
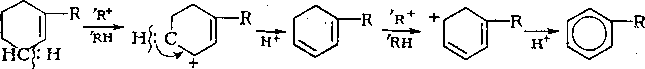
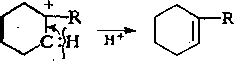
Выход аренов достигает 25 % и более от продуктов превращения циклогексанов, а газы крекинга циклоалканов содержат повышенное по сравнению с газами крекинга алканов количество водорода.
Наблюдается также изомеризация циклогексанов в циклопентаны и обратно:
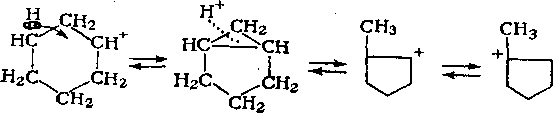
Реакция протекает через протонированное циклопропановое кольцо.
Циклопентаны в условиях каталитического крекинга более устойчивы, чем цнклогексаны. Поэтому’ равновесие сильно сдвинуто вправо. Однако циклогексаны в этих условиях подвергаются дегидрированию в арены. Удаление продукта из сферы реакции смещает равновесие влево. Избирательность превращения циклогексана в бензол или метилциклопентан в конечном счете зависит от катализатора.
При наличии длинных боковых цепей в молекуле циклоал-кана возможны изомеризация боковой цепи и деалкилирование.
Бициклические циклоалканы ароматизируются в большей степени, чем моноциклические. Так, при каталитическом крекинге декалина (500 °С) выход аренов составляет приблизительно 33 % на превращенный декалин. Еще больше ароматических соединений (87,8 %) образуется при крекинге тетралина в тех же условиях.
Превращения алкенов. Скорость каталитического крекинга алкенов на 2—3 порядка выше скорости крекинга соответствующих алканов, что объясняется легкостью образования из алкенов карбкатионов:
Н2С = СНСН2СН3 + Н+ —> НзССНСН2СНз +757 кДж/моль.
При присоединении протона к молекуле алкена образуется такой же ион, как и при отщеплении гидрид-иона от алкана, что определяет общность их реакций при каталитическом крекинге— это изомеризация и Р-распад. Вместе с тем алкенам свойственны также специфические реакции перераспределения водорода и циклизации.
Сущность реакции перераспределения водорода состоит в том, что в присутствии кислотных катализаторов часть алкенов теряет водород и превращается в полиненасыщенные соединения, одновременно другая часть алкенов гидрируется этим водородом, переходя в алканы:
2CsHi6 •—> С.НМ + С8Н18.
Механизм реакции перераспределения водорода можно представить схемой:
СН2 = СНСН2СН2СН2СН2СН2СН3 +Н+ —
—>- СНз СНСН2СН2СН2СН2СН2СНз, СНзСНСН2СН2СН2СН2СН2СНз + сн2 = сн - СН2СН2СН2СН2СН2СН3 —> —> |С8Н)8| + сн2 = сн - СНСН2СН2СН2СН2СН3,
СН2 = СН- СНСН2СН2СН2СН2СН3 —>
—* 1 сн2 = сн — сн = сн - сн2сн1сна~снТ1 + н+.
Алкены, адсорбированные на катализаторе, постепенно теряют водород. Сильноненасыщенные углеводороды полимери-зуются, циклизуются и, постепенно обедняясь водородом, превращаются в кокс.
Циклизация алкенов может привести к образованию цикло-пентанов, циклопентенов и аренов. Например:
СН3СН2СН2СН2СН = СН2
j-Н,
2Hj -ЗН,
v

СНз i^i-СНз l^J-СНз (Oj
Реакция протекает по следующему механизму:
Н R+ +
СН3СН2СН2СНСН=СН2 —> СН3СН2СН2СНСН = СН2 —>
RH н+
я+ +
сн3сн2сн = снсн= сн2 —> СН3СНСН = СНСН = СН2
RH
Н3С j г. rh НзС | || Н3С I. || НзС , ц.
+
Пятичленные циклы изомеризуются в шестичленные и также ароматизируются.
Превращения аренов. Незамещенные арены в условиях каталитического крекинга устойчивы. Метилзамещенные арены реагируют со скоростью, близкой к алканам. Алкилпроизводные аренов, содержащие два и более атомов углерода в цепи, крекируются примерно с такой же скоростью, что и алкены. Основной реакцией алкилпроизводных аренов является деалкилиро-вание. Это объясняется большим сродством ароматического кольца к протону, чем к алкильному иону:
н+
+.?hzch2r
ch2ch2r + Н+ —»- ((jjVjeH2CH2R
CHjCHjR —* |CH2 = CHR | + H+
Скорость реакции возрастает с увеличением длины цепи алкильного заместителя, а также в ряду: ¦ С6Н5 — Сперв С < СбН5 — Свтор < С6Н5 — СТрет, что обусловлено большой устойчивостью образующихся карбкатионов.
В случае метилзамещенных аренов отщепление карбкатиона энергетически затруднено, поэтому в основном протекают реакции диспропорционирорания
2СвН5СН3
< >! СвН„
СвН4 (СНз)э
СНз НзС\
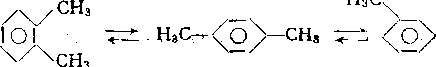
СНз,
и изомеризации по положению заместителей
Полициклические арены прочно сорбируются на катализаторе и подвергаются постепенной деструкции и перераспределению водорода с образованием кокса.
Итак, кокс, образующийся на поверхности катализатора, является смесью сильноненасыщенных полимерных смолообраз-ных алкенов и полициклических аренов. Он блокирует активные центры катализатора и снижает его активность. Для удаления кокса катализатор периодически подвергают регенерации путем окисления.
Сравнение механизмов процессов термического и каталитического крекинга приведено в табл. 13.2.
Таблица 13.2. Сравнение механизмов превращений углеводородов при
термическом и каталитическом крекинге
Каталитический крекинг
Углеводороды
Термические процессы
Алканы Расщепление по С—С-связям
(в газах преобладают углеводороды С[—Сг)
Дегидрирование низших .алканов
Алкеиы Расщепление по С—С-связям
Дегидрирование Полимеризация Диеновый синтез
Диены Диеновый синтез
Цикло- Расщепление по С—С-связям
алканы Дегидрирование
Арены Конденсация с образованием
многоядерных углеводородов Расщепление алкилбеизолов по p-связям С—С
Расщепление по С—С-связям (в газах преобладают углеводороды Q—С4)
Дегидрирование
Изомеризация
Расщепление по С—С-связям
Дегидрирование
Изомеризация
Перераспределение водорода Циклизация
Полимеризация с перераспределением водорода и образованием смолы (кокса) и алкаиов Расщепление по С—С-связям Дегидрирование Изомеризация
Расщепление алкилбензолов по a-связям С—С Изомеризация
Катализаторы процесса и альтернативный механизм реакции. Современные катализаторы крекинга представляют собой сложные системы, состоящие из 10—25 % цеолита Y в редкоземельной или декатионированной форме, равномерно распределенного в аморфном- алюмосиликате, и сформованные в виде микросфер или шариков.
Цеолиты — кристаллические алюмосиликаты — получают обычно кристаллизацией натриевых алюмокремнегелей. Общая эмпирическая формула цеолита может быть записана в виде:
М2/„0 • А1203. jcSi02, где п — валентность металла М; х для цеолита Y равно 3,1—6,0.
Структура цеолита образована тетраэдрами Si04 и AIO4. Атомы алюминия несут одиночный отрицательный заряд, который компенсируется находящимися в пустотах кристаллической решетки катионами металла. Цеолиты с одновалентными катионами неактивны, так как такие катионы полностью компенсируют заряд тетраэдра АЮ4. Замена одновалентного катиона на двух- или трехвалентный приводит к декомпенсации зарядов и создает высокую напряженность электростатического поля, достаточную для образования карбкатионов в результате смещения электронной пары:
С-Н —> С+-Н-, С = С —v С--С+.
Аморфный алюмосиликат, в котором распределен цеолит, обладает собственной активностью. Каталитически активными центрами алюмосиликатов являются как кислоты Бренстеда, так и Льюиса. В качестве кислоты Бренстеда может выступать протон, образующийся из воды, хемосорбированной координационно ненасыщенным атомом алюминия (а), протон гидроксильной группы, связанной с атомом алюминия (б) или кремния (в):
Апротонными кислотными центрами могут быть структуры типа
О-
—О—Si—О—А1—О—.
Переход протонной кислотности в апротонную может быть описан схемой:
О
—О—А1-—О-Н Н+ —» Н20 + —О—А1
О
Наибольшее значение имеют протонодонорные центры, так как полностью дегидратированный алюмосиликат практически неактивен. В цеолитсодержащих алюмосиликатных катализаторах роль катиона металла, по-видимому, состоит в увеличении подвижности протона и стабильности кислотных центров Бренстеда, а также создании дополнительного количества кислотных центров протонизацией молекул воды:
Мега+ ... ОН" ... Н+.
Вследствие этого скорость реакций на цеолитсодержащем катализаторе на 2—3 порядка выше, чем на аморфном. В то же время цеолитсодержащие катализаторы обладают более высокой термической и механической стабильностью, чем чистые цеолиты.
Качественная сторона карбкатионной теории получила общее признание. Однако на ее основе не удается предсказать количественный выход продуктов даже при крекинге индивидуальных соединений. Следует отметить, что существование карбкатионов на поверхности алюмосиликатного катализатора не доказано экспериментально. Возможно, что промежуточными частицами при каталитическом крекинге являются не карб-катионы (a-комплексы), для образования* которых необходим полный гетеролитический разрыв связей, а поверхностные комплексные соединения углеводородов с активными центрами катализатора. Такими соединениями могут быть я-комплексы, для образования которых требуется меньше энергии, чем для образования а-комплексов:
R—НС=СН—R R— НС==СН—R
—А1—
Далее катион-радикалы реагируют по схеме: 0.,
/СНз + СН
^СНз
/СН3
•сн
4 СНз
И
н
А1
А1
\
/
t
А1 +
/Г\
|
сн2=снсн3 | |
|
О | |
Макрокинетика процесса. Каталитический крекинг, как любой гетерогенный каталитический процесс, протекает в несколько стадий: сырье поступает к поверхности катализатора (внешняя диффузия), проникает в поры катализатора (внутренняя диффузия), хемосорбируется на активных центрах катализатора и вступает в химические реакции. Далее, происходит десорбция продуктов крекинга и непрореагировавшего сырья с поверхности, диффузия его из пор катализатора и удаление продуктов крекинга из зоны реакции.
Скорость процесса определяет наиболее медленная стадия. Если процесс протекает в диффузионной области, то скорость его мало зависит от температуры. Для увеличения скорости необходимо применять крупнопористый или сильноизмельчен-ный, например пылевидный, катализатор, что позволит увеличить поверхность катализатора.
Если наиболее медленной стадией является химическая реакция, то скорость процесса зависит главным образом от температуры. Однако увеличивать скорость повышением температуры можно только до определенного предела, после которого реакция переходит в диффузионную область.
Для крекинга нефтяных фракций практически невозможно описать все химические реакции. Поэтому обычно ограничиваются рассмотрением схем, учитывающих основные направления и результирующий эффект крекинга. Кинетику крекинга нефтяных фракций на цеолитсодержащем катализаторе в большинстве случаев представляют уравнением первого порядка: где &Эф — эффективная константа скорости реакции, моль/(с-г); v0 — скорость подачи жидкого сырья, моль/(с-г); X — конверсия сырья, молярные доли.
Более точное описание кинетики каталитического крекинга нефтяных фракций достигается при использовании уравнений, учитывающих дезактивацию катализатора в ходе реакции. Ско-ррсть процесса и выход продуктов крекинга существенно меняются в зависимости от качества сырья, свойств катализатора и полноты его регенерации, технологического режима и конструктивных особенностей реакционных аппаратов.
Каталитический крекинг в промышленности. Каталитический крекинг на алюмосиликатных катализаторах — один из самых многотоннажных процессов в нефтеперерабатывающей промышленности. Целевым назначением процесса является получение высокооктанового бензцна из вакуумных дистиллятов различных нефтей, выкипающих в пределах 300—500 °С.
Каталитический крекинг на цеолитсодержащих катализаторах проводят при 450—530 °С под давлением, близким к атмосферному (0,07—0,3 МПа).
Кроме высокооктанового бензина на установках каталитического крекинга получают также углеводородный газ, легкий и тяжелый газойли. Количество и качество продуктов зависят от характеристики перерабатываемого сырья, катализатора, а также режима процесса.
Ниже представлен материальный баланс установок каталитического крекинга на цеолитсодержащих катализаторах (сырье — вакуумный дистиллят сернистой нефти I, то же после гидроочистки II):
Выход продуктов. %
Углеводородный газ содержит 75—90 % фракции Сз—С*. Его используют после разделения в процессах алкилирования, полимеризации, для производства этилена, пропилена, бутадиена, изопрена, полиизобутилена, ПАВ и других нефтехимических продуктов. Бензиновую фракцию (к. к. 195 °С) применяют как базовый компонент автомобильного бензина. Она содержит аренов 25—40, алкенов 15—30, циклоалканов 2—10 и алканов, преимущественно изостроения, 35—60 % (масс.). Октановое число фракции составляет 78—85 (по моторному методу).
Компоненты, выкипающие выше Ш5 °С, разделяются на фракции. При работе по топливному варианту: 195—350 °С — легкий газойль и >350 °С — тяжелый газойль; при работе по нефтехимическому варианту: 195—270 °С, 270—420 °С и остаток >420 °С. Легкий газойль (195—350 °С) используют как компонент дизельного топлива и в качестве разбавителя при получении мазутов.. Цетановое число легкого каталитического газойля, полученного из парафинового сырья, 45—56, из наф-тено-ароматического — 25—35. Фракцию 195—270 °С применяют как флотореагент, фракцию 270—420 °С — как сырье Для производства технического углерода. Остаточные продукты (>350 °С или >420°С) используют как компоненты котельного топлива или сырья для процессов термического крекинга и коксования.
13.3. КАТАЛИТИЧЕСКИЙ РИФОРМИНГ
Каталитический риформинг — процесс, предназначенный для повышения детонационной стойкости бензинов и получения аренов, главным образом бензола, толуола и ксилолов. Процесс осуществляют при повышенной температуре (около 500 °С) под давлением водорода (1,5—4 МПа) на бифункциональном катализаторе, сочетающем кислотную и гидрирующе-дегидри-рующую функции. Кислотную функцию обычно выполняет оксид алюминия, гидрирующе-дегидрирующую—металлы VIII группы, главным образом платина.
Химические основы процесса. В основе каталитического риформинга лежат три типа реакций:
1)ароматизация исходного сырья путем дегидроциклизации алканов, дегидроизомеризации алкилциклопентанов, дегидрирования циклогексанов;
2) изомеризация углеводородов;
3) гидрокрекинг.
Как и при каталитическом крекинге, осуществление всех названных реакций риформинга ведет к увеличению октанового числа бензина (см. табл. 13.1).
Использование бифункционального катализатора значительно облегчает образование карбкатионов в процессе риформинга по сравнению с каталитическим крекингом, так как необходимые для начала реакции алкены образуются при частичном дегидрировании алканов и циклоалканов на платиновом катализаторе. Алксиы далее протонизируются на кислотном катализаторе и вступают но все реакции, характерные для карбкатио-пов. Поэтому скорость кислотно-каталитических реакций в процессе риформинга выше, чем при каталитическом крекинге.
Превращения алканов. При риформинге алканы подвергаются изомеризации, дегидроциклизации и гидрокрекингу.
Изомеризация алканов протекает по карбкатионному механизму с образованием малоразветвленных изомеров, наиболее термодинамически стабильных в условиях риформинга. Скорость изомеризации возрастает с увеличением молекулярной массы алкана.
Дегидроциклизация — одна из важнейших реакций риформинга, заключающаяся в превращении алканов в арены:
![]()
Дегидроциклизация протекает с поглощением теплоты (около 250 кДж/моль), поэтому константа равновесия реакции возрастает с повышением температуры. Давление сдвигает равновесие реакции влево, т. е. в сторону гидрирования аренов. Однако на практике для уменьшения отложений кокса на катализаторе процесс проводят под повышенным давлением водорода. При температуре 500 °С под давлением водорода 1,5— 1,7 МПа равновесная степень конверсии н-гептана в толуол составляет 95 %.
Механизм ароматизации алканов окончательно не ясен. Возможны следующие пути:
1. Дегидрирование алканов на платине до триена с последующей циклизацией на платине или оксиде алюминия:
С-С —С —С —С-С —> С = С — С = С — С = С —> (о)
— ЗН2 _н, \/
2. Св-циклизация на платине через циклический переходный комплекс
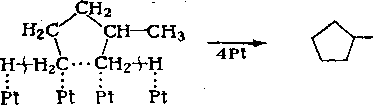
СНз + Н 2
3. Дегидрирование алканов в алкены на платине и циклизация алкенов на оксиде алюминия также с ‘образованием пятичленного цикла. Реакция протекает по согласованному механизму, включающему протонирование двойной связи кислотным центром и одновременный отрыв протона от атома углерода в цепи:
Образовавшиеся пятичленные циклы изомеризуются на кислотных центрах в шестичленные и далее дегидрируются на металле в арены.
Экспериментальные данные свидетельствуют о том, что ароматизация идет по всем рассмотренным направлениям.
Если исходный алкан содержит менее шести атомов углерода в основной цепи, то ароматизации предшествует изомеризация алкана с удлинением основной цепи. Скорость ароматизации возрастает с увеличением длины цепи алкана. Алканы, содержащие десять и более атомов углерода, образуют арены с конденсированными кольцами. Арены с достаточно длинными боковыми цепями могут замыкать дополнительные циклы:
СН2
\СН2
\/ /сн2 знг
СНз
В результате дегидроциклизации алканов образуются гомологи бензола и нафталина с максимальным содержанием метильных заместителей в ядре, которое допускается строением исходного алкана.
Гидрокрекинг алканов приводит к образованию низкомолекулярных соединений:
СН3СН2СН2СН2СН2СН3 СНзСН (СНз) СНз + СНзСНз.
I—> 2СНзСН2СНз
Реакция протекает по следующим стадиям:
- м . , к . Втовичный к
Алкан--* Алкен--> л -->
дегидриро- протони- карокатион изомериза-
ваиие роваиие ция
Изоалкен н ~ л к карбкатион к
—.> Третичный _ Ме„ьшей мо- --
карбкатион (j-распад лекулярной Гротом к™6
Массы тализатору
Изоалкеи и алкеи м Изоалкан и алкан
-*• меньшей молеку---> меньшей молекуляр-
лярной массы гидрирование иой массы
где М—металлический активный центр; К — кислотный активный центр.
Роль гидрокрекинга в процессе риформинга не однозначна. С одной стороны, снижение молекулярной массы алканов приводит к увеличению октанового числа, а с другой стороны, в результате гидрокрекинга образуется значительное количество газообразных продуктов, что снижает выход бензина. Таким образом, роль гидрокрекинга должна быть ограничена. Ниже представлены результаты риформинга «-гексана в зависимости
от температуры при 0,7 МПа и объемной скорости подачи сырья 2 ч-1:
474 °С 500 °С 525 °С
Степень превращения, % 80,2 86,8 90,4
Выход в расчете на превращенный алкан, %
(мол.) аренов (бензол) продуктов изомеризации
| 16,6 | 24,1 |
27,4 |
| 58,0 |
36,9 | 23,4 |
|
25,0 | 38,0 | 49,0 |
продуктов гидрокрекинга
Для снижения роли гидрокрекинга процесс целесообразно проводить при возможно более низком давлении, что одновременно ведет к увеличению равновесного выхода аренов. Результаты рифор-минга н-нонана при температуре 510 °С, объемной скорости 1,5 ч-1 и различном давлении (в % на исходный нонан):
| 0,7 МПа | 2,1 МПа |
|
| Ci—С4 | 10,5 |
21,5 |
|
Неароматические С5 и выше Арены: | 19,0 | 20,0 |
| с6 |
1,6 | 2,0 |
|
с7 | 3,1 |
5,8 |
| с8 |
6*2 | 10,1 |
|
с9 | 54,6 |
36,4 |
Превращения циклоалканов. В условиях риформинга циклоалканы также подвергаются изомеризации, дегидрированию до аренов и гидрокрекингу.
Шестичленные циклоалканы изомеризуются в пятичленные по карбкатионному механизму:
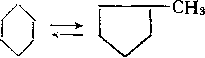
Хотя равновесие изомеризации, как и.при каталитическом крекинге, почти нацело смещено вправо, реакция обратима, так как шестичленные циклоалканы в условиях риформинга дегидрируются в арены, причем равновесие сильно сдвинуто в сторону аренов:
/\ м к I р-СНз
Избирательность превращения циклогексана в метилцикло-пентан и бензол в конечном счете определяется соотношением скоростей реакций и зависит от активности компонентов катализатора. Изомеризация протекает на кислотных центрах по карбкатионному механизму, поэтому при высокой кислотности катализатора будет увеличиваться выход метйлциклопентана (см. разд. 13.2). Дегидрирование происходит на металлическом компоненте катализатора, и с увеличением активности металла будет возрастать скорость образования бензола. Адсорбция шестичленного циклоалкана на металле может сопровождаться либо одновременной диссоциацией шести связей С—Н, либо последовательным быстрым отщеплением атомов водорода:
Реакция эндотермична, поэтому с повышением температуры равновесный выход аренов увеличивается. Скорость дегидрирования гомологов циклогексана выше, чем циклогексана. гем-Замещенные циклогексаиы ароматизируются с отщеплением ме-тильной группы или с ее миграцией:
СНз СНз
/\/ /\_сн.
2l JVh —* IOI + I О I + СН4.
Бициклические щестичленные циклоалканы дегидрируются так же легко, как моноциклические, образуя производные нафталина.
Гидрокрекинг шестичленных циклоалканов происходит в незначительной степени по схеме, описанной для алканов. В условиях риформинга скорость дегидрирования шестичленных циклоалканов в арены значительно выше скорости других реакций (изомеризации в пятичленные и гидрокрекинга). Поэтому селективность превращения циклоалканов в арены составляет практически 100 %.
Пятичленные замещенные циклоалканы в условиях риформинга вступают в следующие реакции:
1. Изомеризация по положению заместителей (через промежуточные карбкатионы):
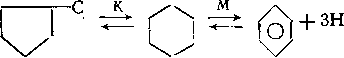
Первая реакция протекает на кислотных центрах катализатора, вторая — на металлических. Выход бензола возрастает с повышением температуры и снижением давления. При температуре 500 °С снижение давления с 3,6 до 1,5 МПа приводит к увеличению выхода бензола с 45 до 90 % (масс.). Дегидрирование циклопентана в циклопентен и циклопентадиен йракти-чески не идет, так как скорость этой реакции значительно ниже скорости дегидроизомеризации. Циклопентадиен прочно адсорбируется на металле и отравляет катализатор.
3. Раскрытие кольца (гидрокрекинг):
СН3 - СН2 - СН2 — СН2 - СН2 — СН3 (1) СНз
СНз-СН -СН2-СН2-СНз СНз
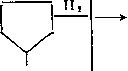
(2)
(3)
СНз —> СНз — сн2- СН - СН2 - СНз.
Относительная скорость гидрогенолиза различных С—С-свя-зей зависит от строения исходного углеводорода, свойств катализатора и условий реакции. На свежем алюмоплатиновом катализаторе гидрогенолиз идет на платине, и соотношение продуктов по реакциям (1), (2) и (3) равно 2,4 : 2,1 : 1. В условиях процесса происходит частичная дезактивация платины, и гидрогенолиз далее протекает на кислотных центрах по карб-катионному механизму:
СНз
СНз
I +
СНз
![]()
|Нг; М
CeHu
Главным продуктом реакции становится и-гексан.
Скорость реакции дегидроизомеризации метилциклопентанов выше, чем изомеризации и гидрокрекинга, поэтому выход бензола при риформинге метилциклопентана достигает 60—70%.
Превращения аренов. Незамещенные соединения в условиях процесса риформинга устойчивы. Метилзамещенные арены (толуол, ксилолы) подвергаются диспропорционированию или изомеризации по положению заместителей. По современным представлениям, изомеризация ксилолов протекает через образование карбкатионов, обусловленное деформацией я-электронного
облака:
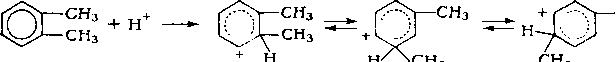
н ХСН3 сн3
![]()
•сн3
СНз
л-Электроны оказывают стабилизирующее действие, и перегруппировки алкилароматических карбкатионов происходят с меньшей скоростью, чем алифатических. Алкиларены, содержащие в боковой цепи 3 и более атомов углерода, деалкили-руются на кислотных центрах по схеме, аналогичной каталитическому крекингу, с последующим гидрированием выделяющегося алкена на металле. В отличие от каталитического крекинга, в условиях риформинга на металлическом катализаторе происходит также деалкилирование метилзамещенных аренов. В результате образуются метан и бензол.
Соединения, содержащие гетероатомы N, S, О и металлы (Pb, As, Си). Эти соединения необратимо сорбируются на платиновом катализаторе и быстро отравляют его. Поэтому присутствие гетероатомных соединений в сырье риформинга нежелательно: содержание серы должно быть не более 1 мг/кг; азота—до 0,5 мг/кг; концентрация Pb, As, Си не должна превышать нескольких миллиграммов на тонну. Для удаления ге-тероорганических и металлорганических соединений сырье риформинга предварительно подвергают гидроочистке.
С наибольшей скоростью идут реакции дегидрирования шестичленных циклоалканов в арены, изомеризации н-алканов в изоалканы и метилциклопентанов в циклогексаны. Наиболее медленно протекают дегидроциклизация и гидрокрекинг алканов (табл. 13.3).
Коксообразование на катализаторе. Закоксовывание катализатора снижает его активность. Механизм образования кокса изучен недостаточно. На платине при умеренных температурах (<427 °С) кокс обр!азуется, по-видимому, в результате диссоциативной адсорбции углеводородов по следующей схеме:
СНзСНз
СНз
СНз
/\
100,
10
10
5
3
13
13
3
4
Таблица 13.3. Относительные скорости и тепловые эффекты реакций каталитического риформинга
|
Относительная скорость | |||
| Тип реакции |
превращения углеводородов |
ля. | |
| с. |
СV | кДж/моль | |
120
+221
-4,6
—15,6
—43,9
—56,4 на моль Hj
+260
Поверхностные соединения обеднены водородом (Н:С=» = 1,0 -т- 1,5), прочно удерживаются на поверхности и находятся в квазиравновесии с газофазным водородом. При более высокой температуре (>477 °С) и атмосферном давлении происходит диссоциация связей С—С, и на поверхности металла образуется углерод. Возможность образования углерода на катализаторе в нормальных условиях риформинга не очевидна, так как процесс осуществляется под давлением водорода. Однако существование поверхностных соединений в реальных условиях риформинга доказано экспериментально.
Модифицированный оксид алюминия по характеру действия аналогичен алюмосиликатному катализатору каталитического крекинга, хотя и обнаруживает меньшую, активность. Коксооб-разование на кислотных центрах катализатора риформинга, как и при крекинге, протекает за счет полимеризации, перераспределения водорода, циклизации, конденсации и других реакций непредельных и ароматических соединений. Образовавшийся кокс состоит из полициклических ароматических колец, связанных с алкеновыми и циклоалкановыми фрагментами.
При всех отличиях механизма коксообразования на платине и оксиде алюминия действие их взаимосвязано: ненасыщенные углеводороды, образующиеся на платине, служат источником кокса на АЬОя. Углеродистые отложения с платины могут ми-
грировать на АЬОз. С другой стороны, продукты уплотнения, в частности полициклические арены, образующиеся на кислотных центрах, достаточно подвижны и могут блокировать металлические центры катализатора. Таким образом, на коксообра-зование влияют обе функции катализатора. Степень дезактивации катализатора зависит от закоксованности как платины, так и AI2O3, так как важнейшие реакции риформинга протекают по бифункциональному механизму.
Катализаторы процесса. Наиболее широкое распространение получили бифункциональные алюмоплатиновые катализаторы, в которых платина в тонкодисперсном состоянии нанесена на оксид алюминия. Платина активна в реакциях гидрирования и дегидрирования. Она способствует образованию аренов и гидрированию промежуточных алкенов. Содержание платины в катализаторе составляет обычно 0,3—0,65 %. Увеличение концентрации платины повышает активность катализатора и октановое число бензина. Однако чрезмерно высокое содержание платины нежелательно, так как при этом усиливается роль реакций деметилирования аренов и расщепления циклоалканов, уменьшающих выход бензина. Основной причиной дезактивации катализатора является его закоксовывание, поэтому повышение стабильности достигается в основном введением модифицирующих добавок, влияющих на коксообразование.
Прогресс каталитического риформинга в последние годы был связан с разработкой платинорениевых катализаторов, содержащих 0,3—0,6 % платины и 0,3—0,4 % рения. Рений образует сплав с платиной и препятствует ее дезактивации, снижая коксообразование путем гидрирования алкенов. Применение биметаллических катализаторов позволило снизить давление риформинга от 3,5 до 1,5—2,0 МПа и увеличить выход бензина с октановым числом 95 пунктов (по исследовательскому методу) примерно на 6 %.
Дальнейшее совершенствование процесса риформинга происходит путем создания полиметаллических катализаторов, со: держащих кроме рения добавки иридия, германия, олова, свинца и других металлов, а также редкоземельных элементов— лантана, церия, неодима. Действие иридия во многом аналогично действию рения. Германий, олово, свинец каталитически неактивны, их используют для подавления активности катализатора в реакциях гидрогенолиза (деметилирования аренов, расщепления циклоалканов), т. е. они играют роль селективного яда. Ранее с той же целью производилось дозированное отравление катализатора серой. Полиметаллические катализаторы обладают стабильностью биметаллических, но характеризуются лучшей избирательностью и обеспечивают более высокий выход бензина. Срок службы полиметаллических катализаторов составляет 6—7 лет. Вместе с тем реализация преимуществ полиметаллических катализаторов требует более тщательной подготовки сырья (очистки от ядов, осушки и пр.).
Кислотную функцию катализатора риформинга выполняет оксид алюминия. Он определяет активность катализатора в реакциях изомеризации, гидрокрекинга и дегидроциклизации. Кислотность поверхности AI2O3 обусловлена как льюисовскими, так и бренстедовскими центрами. Бренстедовская кислотность определяется протонами хемосорбированной воды или протоно-донорных ОН-групп. При частичном удалении ОН-групп путем прокаливания на поверхности остаются координационно-ненасыщенные ионы А13+, которые являются кислотными центрами Льюиса. Для усиления кислотности в оксид алюминия вводят 0,5—2 % хлора. Хлор замещает часть гидроксильных групп поверхности, и катион А13+ оказывается связанным с двумя различными анионами. При этом нарушается электронная симметрия и происходит отток электронов от связи О—Н, благодаря чему повышается подвижность водорода. В процессе работы часть хлора теряется, в основном, за счет взаимодействия с влагой, содержащейся в сырье. Поэтому одно из требований к сырью риформинга — содержание воды не более 10~3 %. Для компенсации возможных потерь хлора в сырье постоянно или периодически вводят определенное количество органических хлоридов (дихлорэтан, четыреххлористый углерод или этил-хлорид).
При длительной эксплуатации катализатора происходят спекание и укрупнецие кристаллов платины от 5—7 до 20 нм. Одновременно снижается и удельная поверхность носителя (АЬОз) от 120 до 83 м2/г, что уменьшает число активных центров. Высокая дисперсность платины поддерживается добавками хлора, образующего комплексное соединение с платиной и оксидом алюминия, а также некоторыми металлическими промоторами. Разрабатывают катализаторы менее требовательные к содержанию в сырье серы, азота, воды, в которых платина введена в цеолит. Регенерацию катализатора осуществляют выжигом кокса с последующим окислительным хлорированием.
Макрокинетика процесса. Характерной особенностью всех модификаций риформинга является то, что одна из его основных стадий — ароматизация — эндотермична, а другая — гидрокрекинг— экзотермична. Результирующий эффект зависит от соотношения удельных интенсивностей обеих этих стадий. По-' вышение температуры способствует ускорению реакций ароматизации и гидрокрекинга. Выход аренов, а следовательно, октановое число бензина при этом возрастают. Вместе с тем в процессе гидрокрекинга образуется много легких углеводородов (С:,—С /,), что приводит к уменьшению выхода бензина. Кроме того, из-за большого расхода водорода в реакциях гидрокрекинга снижается содержание водорода в циркулирующем
Рис. 13.1. Схема превращения углеводородов в условиях процесса риформинга
газе, вследствие чего ускоряется закоксовывание катализатора. В результате наложения этих факторов оптимальная температура проведения процесса составляет 480—530 СС.
Основные реакции риформинга являются типичными реакциями 1-го порядка. Математическое описание риформинга на разных катализаторах должно быть одним и тем же, но с различным численным значением постоянных, присущих каждому катализатору. Для моделирования принята следующая схема риформинга (рис. 13,1).
По данным работы промышленных установок риформинга кажущаяся энергия активации реакции ароматизации составляет 92—158, а гидрокрекинга 117—220 кДж/моль.
С увеличением объемной скорости преобладающую роль в процессе риформинга начинают играть быстроидущие реакции дегидрирования циклоалканов, гидрокрекинга тяжелых алканов и изомеризации углеводородов. Роль реакций дегидроциклиза-ции алканов, деалкилирования аренов и гидрокрекинга легких углеводородов снижается. В результате изменения соотношения между различными реакциями выход бензина возрастает, но его октановое число уменьшается.
Ниже представлена зависимость выхода и свойств бензина каталитического риформинга от объемной скорости процесса:
Объемная скорость, ч~' 2 3 4
Выход дсиропаиизопаниого 91,8 93,9 95,1
бсн:шна, %
Октапопог число бензина (по иселсдоватгльскому методу)
Об-ьсмнор содержание аренов,
%
85,0
81,0
40.6
76.0
38.0
43.0
В промышленных условиях объемную скорость поддерживают на уровне 1—3 ч-1 в зависимости от состава сырья и назначения процесса.
Каталитический риформинг в промышленности. Риформинг в промышленности используют для повышения октанового числа бензиновых фракций и для получения индивидуальных аренов, являющихся ценным сырьем для нефтехимического синтеза.
Процесс осуществляют в среде всдородсодержащего газа (70—90 % (об.) Н2, остальное —низшие углеводороды) при следующих условиях: температура 520—540 °С, давление 1,5—
4 МПа, объемная скорость подачи сырья 1—3 ч"1, отношение количества циркулирующего водородсодержащего газа к сырью 1000—1800 м3/м3.
В качестве сырья для каталитического риформинга обычно используют бензиновые фракции первично! перегонки нефти. В сырье риформинга могут вовлекаться после глубокой очистки бензины вторичных процессов (термического крекинга, коксования, каталитического и гидрокрекинга). Фракционный состав сырья риформинга зависит от назначения процесса. Если целью процесса является получение аренов (бензола, толуола, ксилолов), то используют фракции, содержащие углеводороды Cff (62—85°С), С7 (85—105 °С) и С8 (105—140 °С). Если процесс проводят с целью получения высокооктанового бензина, то сырьем служит фракция 85—180 °С, соответствующая углеводородам С7—Сэ.
Основными продуктами риформинга являются водородсодержащий газ и жидкая фракция (риформат). Водород используют частично для восполнения потерь циркулирующего водородсо-держащего газа. Большую часть водорода направляют на установки гидрокрекинга и гидроочистки нефтепродуктов. Объемный выход технического водорода с содержанием 90 % в процессе риформинга на платинорениевом катализаторе составляет
1,3—2,5 %. Из водородсодержащего газа при стабилизации выделяют сухой (Cj—С2 или Ci—Сз) и сжиженный газы (Сз—С4).
Риформат используют как высокооктановый компонент автомобильных бензинов (октановое число 85 по моторному методу или 95 по исследовательскому) или направляют на выделение аренов.
Бензин каталитического риформинга содержит 50—70 % аренов, около 30 % н- и пзоалканов, 10—15 % циклоалканов и 2 % непредельных соединений. Бензин каталитического риформинга из-за высокого содержания аренов, приводящего к повышенному нагярообразованию, по может в чистом виде использоваться п качестве топлива для автомобилей и подвергается компаундированию.
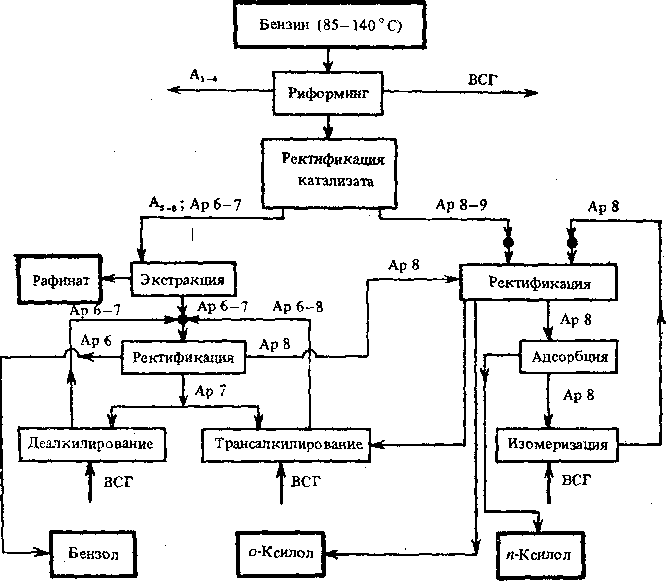
Рис. 13.2. Схема комплексного производства аренов
Л1_4- алкаиы Ci—С*; As_7—алкаиы Св—Се; Арб— бензол; Ар7—толуол; Ар8—9 —
арены С8—Сэ; ВСГ — водородсодержащий газ; •—очистка активной глиной
Из бензинов каталитического риформинга можно выделить индивидуальные арены. Деароматизированную часть катализа-тов — рафинаты — используют как компонент бензина и сырье пиролиза, а также для получения легких парафиновых растворителей.
Наибольшее значение в качестве нефтехимических продуктов и аренов приобрели бензол, о- и п-ксилолы. Для разделения аренов, а также для превращения риформатов в наиболее ценные продукты созданы комплексы производства. Один из вариантой такого комплекса представлен на рис. 13.2.
Головной процесс в таком комплексе — обычно каталитический риформинг. При проведении процесса в жестком режиме получают риформат, содержащий около 70 % аренов. Из них бензола — 3,0%, толуола — 22,0%, ксилолов — 35,0%, ареной Св—Сю — 9,5 %. Риформат делят на две фракции: легкую и тяжелую. Легкая содержит алканы Cs—Cs и арены (бензол и толуол); тяжелая — главным образом арены Са—Сэ.
Легкую фракцию экстрагируют сульфоланом. Полученный экстракт вместе с продуктами деалкилирования и трансалки-лирования подвергают очистке от алкенов на активной глине при 170—180 °С под давлением 0,7—1,0 МПа. Очищенный продукт разделяют ректификацией на товарный бензол, толуол,, ксилолы. Часть толуола поступает на секцию термического гид-родеалкилирования, где при 670—760 °С под давлением водорода 3 МПа получают бензол:
С6Н5СН3 -—> С6Н6 -f- СН4.
Для превращения аренов Ст и С9 в бензол и кснлолы в состав комплекса включена секция трансалкилирования. Реакцию проводят на кислотных катализаторах при 500 °С под давлением 3 МПа:
С6Н5СНз + СбНз (СНз)з 2СбН* (СН3)2.
Наряду с трансалкилированием в этих условиях происходит диспропорционирование толуола:
2С6Н5СНз —^ С6Н6 + С6Н4 (СНз)2.
Продукты реакции после очистки на глине поступают на ректификацию.
Тяжелую фракцию риформата (в смеси с продуктами изомеризации) очищают от непредельных углеводородов на активной глине и направляют на четкую ректификацию. Туда же поступает и ксилольная фракция продуктов трансалкилирования. При четкой ректификации выделяют товарный о-ксилол; арены Сд направляют на трансалкилирование, а смесь аренов-Ся (п- и м-ксилолы и этилбензол) — на адсорбцию для выделения n-ксилола. Адсорбция протекает на цеолитах (процесс Па-рекс). Оставшаяся смесь ж-ксилола и этилбензола изомери-зуется на бифункциональном катализаторе, содержащем платину на кислотнбм носителе, при 400—450 °С под давлением
1,4—2,4 МПа. Продукты изомеризации возрращаются на ректификацию.
На комплексной установке мощностью 890 тыс. т/год при каталитическом риформинге фракции 85—140 °С получают: 126 тыс. т/год бензола и по 165 тыс. т/год о- и га-ксилолов.
Ниже представлены источники образования целевых продуктов (в %):
/2-Ксилол
35
Бензол
о-Ксилол
16
52
32
15
30
Риформинг
Деалкнлиропаппс
Трапсалкилнронание
Изомеризация
13.4. СИНТЕЗ ВЫСОКООКТАНОВЫХ КОМПОНЕНТОВ ТОПЛИВ
Для получения высококачественных бензинов с октановым числом по исследовательскому методу 93—98 (АИ-93 и АИ-98) катализат жесткого риформинга с содержанием аренов 65— 70 % требуется разбавлять разветвленными алканами (изокомпонентами) с октановым числом не ниже 85—90 пунктов. Такие продукты получают в процессах изомеризации, алкилирова-лия и полимеризации легких углеводородов и нефтяных газов.
Оптимальное содержание аренов в компаундированных бензинах составляет 35—45 %.
Значение процессов получения изокомпонентов возрастает в связи с жесткими нормами на содержание тетраэтилсвинца.
Изомеризация алканов С4 — С6. Реакции изомеризации имеют широкое распространение в нефтеперерабатывающей промышленности. Их нельзя не учитывать при рассмотрении процессов каталитического крекинга и риформинга; кроме того, онн имеют самостоятельное значение и используются для повышения октановых чисел компонентов моторных топлив и получения индивидуальных изоалканов С4 и С5. Изобутан применяют в процессе алкилирования и для получения изобутилена для синтеза метил-грег-бутилового эфира. Изопентан подвергают дегидрированию с целью получения изопрена для нефтехимической промышленности.
Высокооктановый компонент бензина получают изомеризацией наиболее легкой части бензина прямой перегонки — фракции С4 — Се. Изомеризация высших алканов не дает существенного повышения октанового числа. Вместе с тем слабораз-ветвленные алканы с длинной цепью являются желательными компонентами реактивных и дизельных топлив, а также масляных фракций. Они имеют низкую температуру застывания и хорошие цетановые и вязкостно-температурные характеристики. Изомеризация высокомолекулярных алканов повышает качество топлив и масел и в ряде случаев успешно конкурирует с депа-рафинизацией нефтяных фракций.
Термодинамика, кинетика и механизм процесса. Термодинамическое равновесие в смеси нормальных и разветвленных алканов с повышением температуры смещается в сторону неразветвлеиных и малоразветвленных структур. Изомеризация алканов — процесс экзотермический, но количество выделяющейся теплоты невелико — 6—8 кДж/моль.
Каталитическая изомеризация протекает обычно как реакция первого порядка с кажущейся энергией активации «40 кДж/моль. В присутствии кислотных катализаторов изомеризация протекает по цепному карбкатионному механизму. Первая стадия процесса — образование карбкатиона R+ — опре-
деляется катализатором. Затем следует стадия передачи цепи: R+ + СН3СН2СН2СН3 ^ RH + СНзСНСН2СНз. Следующие две реакции представляют собой звено цепи: СНзСНСН2СНз СНзСН (СНз) СН2 СНзС (СНз)СНз,
СНзС (СНз) СНз + СНзСН2СН2СНз
I СНзСН (СНз) СНз | + СН3СНСН2СН3.
Повторением этого звена осуществляется развитие цепи.. Обрыв цепи происходит вследствие переноса протона от карбка-тиона к катализатору.
В присутствии катализаторов электронного типа (гидрирования— дегидрирования) наиболее вероятен радикальный механизм, допускающий первоначальную диссоциацию молекул. Механизм изомеризации на бифункциональном катализаторе можно представить схемой: вначале происходит дегидрирование нормального алкана на активном центре металлического катализатора (М), затем на кислотном центре (К) образовавшийся алкен превращается в карбкатион, изомеризуется и после передачи протона катализатору выделяется в виде разветвленного алкена. Последний гидрируется на металле:
м К; н+
СНзСН2СН2СНз СН3СН2СН=СН2
н2
—СНзСН2СНСНз СНзС(СНз)СНз
н+
М; Н2
СН2 = С (СНз) СНз 4=* СНзС(СНз)СНз.
На кислотном активном центре катион находится в ионно» паре с анионом. При изомеризации бутана побочные продукты отсутствуют. При изомеризации пентана и алканов с большим числом углеродных атомов получают алканы легче и тяжелее исходного углеводорода, а также алкены и циклоалкены. Образование побочных продуктов объясняется алкилированием промежуточных алкенов, циклизацией, перераспределением во^ дорода и распадом карбкатионов с большим числом атомов углерода.
Катализаторы изомеризации. В ранних модификациях промышленного процесса изомеризация осуществлялась на хлориде или бромиде алюминия с использованием в качестве промоторов алкена и сухого хлористого водорода. Ионы
HCl + AlCh HA1CU,
СНзСН = СНСНз + НА1Си (СНзСН2СНСНз)А1С14-
Процесс протекал при относительно низкой температуре *90—120 °С, и равновесие реакции было сдвинуто в сторону разветвленных алканов. К недостаткам этого процесса относят: высокую коррозионную агрессивность катализатора; трудность отделения углеводородов от катализатора; низкую селективность процесса из-за образования большого количества побочных продуктов; большие потери катализатора в результате его гидролиза и растворимости в углеводородах — около 1 %.
Эти недостатки привели к утрате его промышленного значения. Современные промышленные катализаторы изомеризации алканов представляют собой бифункциональные системы «металл — носитель» типа катализаторов риформинга. В качестве металлического компонента катализатора используют платину или палладий, в качестве носителя — фторированный или хлорированный оксид алюминия, аморфные или кристаллические алюмосиликаты, внесенные в матрицу оксида алюминия. Для предотвращения закоксовывания катализатора процесс проводят под давлением водорода 1,4—4 МПа. Первые алюмо-платиновые катализаторы, содержащие 1—2 % хлора или •фтора, обладали недостаточной активностью, поэтому процесс проводился при высокой температуре (350—400°С), что снижало термодинамически возможную степень изомеризации. Этот процесс в технике получил название высокотемпературной изомеризации. Повышение активности катализатора и снижение рабочих температур до 230—380 °С было достигнуто увеличением кислотности носителя при переходе на металлцеолитные катализаторы (среднетемпературная изомеризация). Наибольшую активность имеют платиновые или палладиевые катализаторы на оксиде алюминия, содержащие 7—10 % хлора. Они позволяют проводить реакцию при температуре 100—200°С (низкотемпературная изомеризация). Необходимым условием изомеризации на бифункциональных катализаторах, как и каталитического риформинга, является глубокая очистка сырья и водородсодержащего газа от примесей влаги, серы, азота и кислорода, отравляющих катализатор. Для восполнения потерь галогена на катализаторе в сырье вводят небольшое количество галогенсодержащих соединений.
В последнее время и качество катализаторов изомеризации предлагаются «сверхкислоты», такие как HF — BF3, HF — SbF5, HS03F — SbFs и ДР- В присутствии этих катализаторов в атмосфере водорода изомеризация алканов протекает быстро при
температуре 20—50 °С. Карбкатион образуется прямо из алкана:
С4Н10+Н+ —> ГСНз-СН-СН2СНзП+ —>
—-> Н2 + СНзСНСНгСНз -
Однако эти катализаторы еще не получили промышленного применения.
Наибольшее значение в настоящее время имеют процессы-низко- и высокотемпературной изомеризации.
Алкилирование изоалканов алкенами. Процесс алкилирования изобутана применяют в нефтеперерабатывающей промышленности с целью получения компонента высокооктанового бензина. Алкилат обладает оптимальным комплексом эксплуатационных свойств. Октановое число основного продукта алкилирования изобутана бутенами — 2,2,4-триметилпентана (изооктана) принято за 100.
Алкилирование разветвленных алканов алкенами в общем виде описывают уравнением:
C/i^2n + 2 CmH2m> Сп + mH2 (га + т) + 2-
Реакция идет с выделением теплоты (85—90 кДж/моль),. поэтому предпочтительно проводить процесс при низкой температуре.
Механизм алкилирования в присутствии кислотных катализаторов—карбкатионный цепной.
На первой стадии алкен (бутен-2) реагирует с протоном катализатора:
СНзСН = СНСНз + Н+А- —> СН3СНСН2СН3 + А-.
Бутильный катион реагирует с разветвленным алканом:
СНзСН (СНзЬ +СН3СНСН2СН3 ¦—у
—* СНзС (СНз)2 + СН3СН2СН2СН3.
Образовавшийся третичный бутильный карбкатион присоединяется к молекуле алкена:
СНзС (СНзЬ + СНзСН = СНСНз - >
- > СНзС (СНзЬ СН (СНз) снсн,.
Вторичный октальный карбкатион изомеризуется в более устойчивый третичный:
СНзС (СНзЬ С (СНз) СН2СНз —>¦ -> СНз+С (СНз) С (СНз)2СН2СНз
СНзС(СНз)2СН (СНз)СНСНз
->¦ СНзС (СНз)2 СНаС (СНз) СНз .
СНзС (СНз) СН (СНз) СН (СНз) СНз
Изомеризованный ион вступает в реакцию с исходным разветвленным алканом с образованием конечных продуктов реакции— изооктанов и новых карбкатионов:
СНзС (СНз) С (СНз)2 СН2СНз + СНзСН (СНз)2 —>
—> СНзСН (СНз) с (СНзЬ СН2СНз + СН3С (CH3)s.
Реакции присоединения третичного карбкатиона к молекуле алкена, изомеризации вторичного октильного карбкатиона и образования конечных продуктов представляют собой звено депи, повторение которого приводит к цепному процессу.
Обрыв цепи происходит при передаче протона от карбкатиона к аниону кислоты:
СНзС (СНз) С (СН3)2СН2СНз + А- —->•
—>- СН2 = С (СНз) С (СНз)2СН2СНз + Н+А".
При алкилировании наблюдается ряд побочных реакций: деструктивное алкилирование, полимеризация алкенов, а также взаимодействие алкенов с катализатором (серной кислотой) с образованием сложных эфиров (сульфатирование алкенов).
Деструктивное алкилирование происходит в результате 0-распада промежуточных карбкатионов и приводит к образованию углеводородов С5—С7. Роль этого процесса уменьшается ¦с понижением температуры. Полимеризация алкенов дает продукты большей молекулярной массы, чем С8. Процессы полимеризации подавляются избытком изобутана.
Алкилирование проводят под давлением, для того чтобы углеводороды находились в жидкой фазе. Процесс является гетерогенным, реакция протекает в эмульсии кислота — углеводород. Кислоты (H2S04; HF), применяемые в качестве катализаторов, малорастворимы в углеводородах, а без противоиона в среде слабополярных углеводородов карбкатионы существовать не могут. Поэтому реакция идет на границе раздела фаз или в среде (в тонкой пленке) кислоты. Процесс алки-лирования лимитируется массопередачей реагентов из углеводородной фазы в кислотную, поэтому важную роль играет интенсивное перемешивание, необходимое для создания однородной эмульсии.
Катализаторы алкилирования. Основное значение для получения высокооктановых алкилатов имеют серная и фтористоводородная кислоты. Для алкилирования обычно применяют серную кислоту (96—98 %). При дальнейшем увеличении концентрации кислоты возрастает роль процессов окисления и сульфатирования углеводородов. Снижение концентрации кислоты уменьшает ее активность в реакции алкилирования,. а так как растворимость алкенов в серной кислоте значительно больше растворимости изобутана, существенно возрастает скорость полимеризации алкенов. Кроме того, разбавление кислоты создает опасность коррозии оборудования. Снижение концентрации (и, следовательно, активности) серной кислоты в ходе процесса обусловлено разбавлением водой, содержащейся в сырье и образующейся за счет частичного окисления углеводородов, и в меньшей степени — продуктами окисления и сульфатирования. Расход серной кислоты в побочных процессах составляет 100—160 кг на 1 т алкилата.
Фтороводород значительно лучше растворяет изобутан, чем серная кислота, поэтому соотношение изобутан: алкен в зоне реакции (в пленке кислоты, в которой идет реакция) значительно выше. Вследствие этого алкилирование протекает практически без побочных реакций и выход основных продуктов .выше, чем при катализе серной кислотой. Катализатор сохраняет высокую активность при содержании в нем воды не более 1,5% и органических разбавителей — не более 12%. Заданную-концентрацию фтороводорода поддерживают за счет отбора части катализатора на регенерацию. Фтороводород легко отделяется от воды перегонкой. Расход HF составляет примерно*
0,7 кг на 1 т алкилата.
Перспективными катализаторами алкилирования являются «сверхкислоты» —комплексы трифторида бора с фтороводоро-дом и серной кислотой.
В СНГ алкилат получают только при катализе серной кислотой.
Сернокислотное алкилирование в промышленности. В присутствии серной кислоты алкилирование проводят при температуре 5—15 °С, давлении 0,3—0,6 МПа* концентрации серной кислоты 88—99%, соотношениях кислота : углеводороды 1,1: 1,5 и изобутан : алкены (6—10) : 1.
Применение низкой температуры обусловлено увеличением равновесного выхода разпетвленных алканов и снижением рол» побочных реакций p-расщепления промежуточных ионон, сульфатирования и окисления. Повышенное давление необходимо для поддержания реакционной, смеси в жидком состоянии. При соотношении кислота : углеводороды (1,1 -f- 1,5) : 1 достигается полное вовлечение углеводородной фазы в эмульсию. Избыток изобутана способствует подавлению процессов полимеризации и увеличению выхода алкилата.
Сырьем установок алкилирования являются изобутан, бу-тан-бутиленовая и пропан-пропиленовая фракции, получаемые преимущественно в процессах каталитического и термического крекинга. Алканы нормального строения С3 — С5 в реакцию алкилирования не вступают и являются инертными примесями; повышение их концентрации в сырье приводит к снижению скорости транспортирования реагирующих веществ, поэтому их содержание должно быть минимальным. Из разветвленных алканов наибольшее значение имеет изобутан. Применение в качестве сырья для алкилирования изопентана нецелесообразно ввиду того, что он является ценным высокооктановым компонентом бензина и сырьем для производства изопрена.
Существенное влияние на показатели процесса оказывает состав алкенов. Этилен практически не алкилирует нзобутан, а главным образом сульфатируется и полимеризуется. Пропилен вступает в реакцию с изобутаном, но октановое число меньше, чем при использовании бутиленов (табл. 13.4). Кроме того, при алкилировании изобутана чистым пропиленом очень высок расход серной кислоты. Высшие алкены (С5 и выше) в процессе реакции образуют ионы большой молекулярной массы, склонные к расщеплению с образованием низкомолекулярных продуктов, что снижает выход алкилата. Таким образом, оптимальным сырьем для алкилирования изобутана являются бу-тилены (см. табл. 13.4).
В промышленных условиях в качестве алкенового сырья обычно используют пропан-пропиленовую фракцию в смеси с бутан-бутиленовой в соотношении, обеспечивающем содержа-
Та блица 13.4. Зависимость показателей процесса алкилирования изобутана от длины цепи алкена
|
Показатели | Сырье |
||
| пропилеи |
бутилен | амилен | |
| Объемный выход алкилата в расчете | 175—187 | 170—172 |
160 |
| на алкены, % | |||
|
Объемный расход изобутана в расче |
127—135 | 111—117 |
90 -140 |
| те на алкеновое сырье, % | |||
| Октановое число алкилата |
|||
| моторный метод | 87—90 | 92—94 | 87—89 |
|
исследовательский метод |
89-91 | 92-96 | 88—90 |
ние пропилена менее 50 % от суммы алкенов С3 и С4. Целевым продуктом является легкий алкилат (к. к. 185 °С), применяемый как высокооктановый компонент бензинов. Выход алкилата в оптимальных условиях составляет 200—220 % от содержания алкенов Сз и С< в сырье. Алкилат имеет октановое число 91—95 (по моторному методу). Кроме легкого алкилата с установки удаляют непрореагировавшие инертные алканы нормального строения С3 — С4, избыточный изобутан, а также тяжелый алкилат (фракция 185—310 °С). Газы Сз — С4 используют как сырье для нефтехимии, тяжелый алкилат — как растворитель для различных целей или компонент дизельного топлива. Материальный баланс алкилирования представлен ниже (сырье — смесь алкан-алкеновых фракций С3 и С4):
| Поступило. % |
Получено, % | |
| Пропан | 6,8 | 6,3 |
| Пропилен | 19,7 |
— |
| я-Бутен |
4,2 | 4,5 |
|
Бутиленьг | 19,8 |
— |
| Изобутан |
49,5 | 2,1 |
|
Легкий алкилат | (к. к. — | 83,1 |
| 195 °С) | 3,0 | |
|
Тяжелый алкилат | (выше — | |
| 195 °С) | ||
| Потери | — |
1,0 |
| В се го | ! 00,0 | 100,0 |
Полимеризация алкенов. Процессы полимеризации пропилен- и бутиленсодержащих фракций, извлекать из которых чистые алкены нецелесообразно, предназначены для получения низкомолекулярных полимеров, используемых для производства моторных топлив и нефтехимического сырья.
Термодинамика, макрокинетика и механизм процесса. Полимеризация алкенов термодинамически возможна (значение свободной энергии Гиббса отрицательно) при температуре не выше 227—277 °С. Реакция экзотермична. Тепловой эффект составляет около 70 кДж на 1 моль полимеризо-вавшегося алкена. Равновесная степень полимеризации возрастает с увеличением давления и снижением температуры.
На кислотных катализаторах реакция протекает по карбка-тионному механизму; при этом алкен находится в газовой фазе и реакция идет на поверхности твердого катализатора или в пленке кислоты. На твердом катализаторе карбкатионы могут существовать только в ионной парс с анионом, входящим в фазу катализатора; в пленке кислоты часть ионных пар может диссоциировать на кинетически независимые ионы.
Механизм реакции может быть описан следующей схемой {на примере пропилена).
1. Протонирование алкена на катализаторе:
. СН2 = СНСНз + НА — v (СН3СНСН3) А-.
2. Присоединение образовавшегося иона к молекуле алкена по я-связи:
СНзСНСНз + СН2 = СНСНз —» СН3СН (СНз) СН2СНСН3.
Гексильный ион может далее присоединить последовательно еще несколько молекул алкена с образованием ионов Сд и С12.
3. Изомеризация ионов — вторичные карбкатионы превращаются в более устойчивые третичные:
СНзСН(СНз)СН2СНСНз СНзСН(СНз)СНСН2СНз ^:СНзСН2С(СНз)СН2СНз
СНзС(СНз)СН2СН2СНз СНзСН(СНз)С(СНз)СНз.
Ионы Сд и С12 также подвергаются изомеризации, однако затем они легко распадаются. Распад ионов Сд и Ci2 приводит к образованию продуктов с числом атомов углерода не кратным трем.
4. Реакция заканчивается передачей протона от карбкатиона катализатору или исходному алкену; возможен также отрыв гидрид-иона от исходного алкена карбкатионом. В последнем случае образуется алкенильный ион, дальнейшие превращения которого ведут к образованию высоконенасыщенных продуктов, прочно связанных с поверхностью катализатора, или циклических углеводородов.
По склонности к полимеризации алкены располагаются в ряд: изобутилен > бутилены > пропилен > этилен.
В промышленных условиях при использовании в качестве катализатора фосфорной кислоты на кизельгуре скорость реакции лимитируется массопередачей. Кажущаяся энергия активации составляет всего 21—31 кДж/моль. Порядок реакции близок к 1, и ее скорость, таким образом, прямо пропорциональна парциальному давлению алкена.
' Температура проведения процесса ие должна быть слишком низкой, так как уже при 130 °С вместо полимеризации идет образование фосфорнокислых эфиров. Нельзя допускать также чрезмерного повышения температуры, так как выше 220 °С увеличивается вероятность распада полимерных карбкатионов. Кроме того, при высокой температуре интенсифицируется реакция отрыва гидрид-иона от исходного алкена, в результате чего увеличивается выход смолообразных ненасыщенных продуктов, блокирующих поверхность катализатора. При температуре выше 270 °С полимеризация становится термодинамически невозможной. Оптимальной является температура 190—230 °С.
Повышение давления не только ускоряет процесс, но и увеличивает срок жизни катализатора, так как удерживает в жидкой фазе олигомерные продукты, которые смывают с поверхности катализатора смолистые отложения. Давление не способствует утяжелению полимербензина, так как при используемых температурах полимерные карбкатионы с высокой молярной массой быстро распадаются, поэтому полимеризацию проводят обычно под давлением 1,7—8,0 МПа.
Для предотвращения побочных реакций процесс ведут не до полного использования алкенов, а обеспечивают примерно 90 %-ю конверсию, регулируя температуру, давление, активность катализатора. В зависимости от этих факторов объемная скорость составляет 1,7—4,0 ч~!.
Катализаторы полимеризации. Полимеризация алкенов С3 — С4 с получением смеси разветвленных алкенов, перегоняющихся в пределах температур кипения бензинов, катализируется разнообразными катализаторами катионной полимеризации. Наибольшее значение имеет фосфорная кислота {Р205 на кизельгуре). Приблизительный состав катализатора — P205-Si02-2H20. Активность катализатора (до 57—64 % Рг05) зависит от содержания воды в сырье (до 0,035—0,040 %) и температуры процесса. При избытке воды происходит уплотнение катализатора и увеличение перепада давления в реакторах. В некоторых зарубежных схемах используют растворы катализаторов (до 1 % на сырье), которые в конце процесса отделяются от продуктов и рециркулируют или нейтрализуются.
Промышленное получение полимербензина. Полимеризацию алкенов с целью получения полимербензина в промышленных условиях осуществляют при температуре 190— 230 °С и давлении около 6 МПа в присутствии фосфорнокислых катализаторов. Сырьем процесса полимеризации являются про-пан-пропиленовая и бутан-бутиленовая фракции (ППФ и ББФ) каталитического крекинга, содержащие 30—37 % алкенов, или пиролиза — с более высокой концентрацией алкенов.
Продукты полимеризации ППФ, главным образом изогек-сены, имеют октановое число 81—84 (по моторному методу) и до 94—97 (по исследовательскому методу). Продукты полимеризации ББФ обладают более высокими октановыми числами: до 85 (по моторному методу) и около 100 (по исследовательскому методу). Сополиконденсаты ППФ и ББФ характеризуются промежуточными значениями октановых чисел. Октановое число, смеси углеводородов не является аддитивной функцией их октановых чисел. Полимербеизин существенно увеличивает порог детонации при добавлении к бензинам другой природы; его октановое число смешения составляет 90—130 (по моторному методу), т. е. в смеси с другими бензинами он ведет себя как компонент с октановым числом 90—-130 (по моторному методу) .
В связи с тенденцией использования бутиленов для производства метил-трет-бутилового эфира, алкилата или вго/?-бути-лового спирта возрастает роль процессов получения высокооктановых компонентов бензина из пропан-пропиленовой фракции.
При производстве сырья для нефтехимии продукцией процесса являются тримеры или тетрамеры пропилена или олигомеры Сз- В качестве побочных продуктов выделяются отработанные фракции газов С3 — С4, а также фракция дизельного топлива.
Сжиженные газы и высокооктановые кислородсодержащие продукты. Сжиженные углеводородные газы имеют высокую ан-тидетонационную стойкость (октановое число 93—100 по исследовательскому методу) и являются полноценным заменителем неэтилированного бензина. Кроме того, сжиженные газы являются экологически чистым топливом: при переходе с бензина на сжиженные газы выбросы оксида углерода снижаются в 5 раз, а выбросы углеводородов — более чем на 50%. Согласно ТУ 38001302—-73 газообразное топливо содержит 75— 85 % пропана и 15—25 % бутанов. Расширение использования газобаллонных автомобилей увеличит целесообразность использования процессов гидрокрекинга для облагораживания бензиновых фракций, так как полученный сжиженный газ будет таким же ценным топливом, как и высококачественный бензин.
В качестве высокооктановых добавок к бензинам применяют также кислородсодержащие вещества, имеющие октановые числа смешения 120—150 пунктов (низшие алифатические спирты, метил-трег-бутиловый эфир).
