Глава 5 методы разделения компонентов нефти и газа
Глава 5
МЕТОДЫ РАЗДЕЛЕНИЯ КОМПОНЕНТОВ НЕФТИ И ГАЗА
5.1. КЛАССИФИКАЦИЯ МЕТОДОВ РАЗДЕЛЕНИЯ
Для облегчения анализа нефтей и нефтепродуктов используют разнообразные методы их предварительного разделения как по молекулярным массам, так и по химическому составу^ Для разделения нефти и выделения различных групп углеводородов и гетероатомных компонентов применяют химические и-физические методы. Химические методы основаны на неодинаковой реакционной способности разделяемых компонентов, а физические (или физико-химические) — на различии концентраций компонентов в сосуществующих равновесных фазах (табл. 5.1).
Простыми условно названы методы разделения, при которых изменение концентрации разделяемых компонентов в сосуществующих фазах достигается лишь благодаря сообщению-системе энергии, а сложными — методы с применением дополнительных разделяющих агентов (селективных растворителей, адсорбентов и т. д.), увеличивающих различие составов; фаз.
К физико-химическим методам разделения относят также разнообразные виды хроматографии, различающиеся агрегатным состоянием подвижной и неподвижной фаз (см. гл. 6).
Сочетание эффективных приемов разделения с современными инструментальными методами анализа позволило создать-информативиые экспресс-методики определения качественного и количественного состава нефтей и нефтепродуктов.
Диффузия через мембра- Диффузия с газом-носи-
Газ — газ
Раз — жидкость
Газ — твердая фаза Жидкость — жидкость
Жидкость — твердая фаза
иу
Перегонка и ректификация
водяным
ректифика-ректифи-
Возгонка
Диффузия через мембрану
Термическая диффузия Кристаллизация
тел ем Перегонка паром Абсорбция Аз еотройная цня
Экстрактивная
кацня
Адсорбция
Экстракция
Адсорбцня
Экстрактивная кристаллизация
Аддуктнвиая кристаллизация
5.2. ПЕРЕГОНКА И РЕКТИФИКАЦИЯ
Перегонка — старейший метод разделения нефти на фракции, содержащие компоненты с близкими молекулярными массами, которым удалось выделить нз нефтей ряд индивидуальных соединений. Так, еще в конце XIX века дробной перегонкой были выделены и идентифицированы пентан, изопентан, 2-ме-тилпентан, 2,3-диметилбутан, 2- и 3-метилгексаны и ряд других низкокипящих углеводородов.
Различные виды перегонки и ректификации широко используют и в настоящее время — ни одна схема анализа нефтей не обходится без фракционирования при атмосферном давлении или под вакуумом.
Фракционный состав нефти определяют с помощью перегонки при атмосферном давлении без ректификации на стандартном аппарате. При этом оценивают выход фракций, выкипающих до 300°С; перегонять более высококипящие нефтяные фракции и нефтепродукты при атмосферном давлении не рекомендуется, так как они при этом могут разлагаться.
Для определения группового углеводородного и структурно-группового состава обезвоженную нефть разделяют ректификацией при атмосферном давлении на унифицированных аппаратах (типа ЦИАТИМ-58а или АРН-2) на стандартные фракции: н. к. —60, 60—95, 95—122, 122—150, 150—200°С. Затем под вакуумом при остаточном давлении 666,5—133,3 Па (5— 1 мм рт. ст.) отбирают средние фракции: 200—250, 250—300 и 300—350 °С. Для приведения температур кипения в вакууме к температурам кипения при атмосферном давлении пользуются специальными пересчетными формулами или номограммами, чаще всего номограммой UOP.
Для фракционирования масляных фракций вместо насадоч-ных аппаратов можно применять колонки с вращающимся ротором, имеющие меньшее гидравлическое сопротивление и обеспечивающие получение фракций без разложения вплоть до 550 °С.
Для выделения высокОкипящих масляных фракций возможно использование молекулярной перегонки. Процесс протекает в глубоком вакууме (остаточное давление <0,1 Па) при небольшом расстоянии между поверхностями испарения и конденсации (10—30 мм), меньшем, чем длина свободного пробега молекул. В связи с этим испарившиеся молекулы не сталкиваются и достигают конденсатора с минимальными затратами энергии. Современные роторные пленочные аппараты позволяют отгонять фракции с температурой кипения до 650°С практически без разложения.
Ректификация при различных давлениях используется для выделения индивидуальных углеводородов из бензиновых фракций. При этом учитывается, что наиболее пологий характер-зависимости давления насыщенного пара от температуры отмечается для н-алканов, более крутая зависимость характерна для алканов изостроения и циклоалканов.
Для разделения смесей углеводородов с близкими температурами кипения, например аренов Се, необходима сверхчеткая ректификация. Так, для выделения наиболее высококипящего изомера — о-ксилола (коэффициент относительной летучести а ключевой пары компонентов л-ксилол — о-ксилол при 180°С равен 1,135) используют ректификационные колонны, имеющие 100—150 тарелок при кратности орошения (5 8): 1.
Одним из методов газоразделения наряду с абсорбцией является низкотемпературная ректификация с использованием таких хладагентов, как аммиак или пропан.
5.3. АЗЕОТРОПНАЯ И ЭКСТРАКТИВНАЯ РЕКТИФИКАЦИЯ,
АБСОРБЦИЯ,ЭКСТРАКЦИЯ
Разделение нефтяных фракций на группы компонентов по химическому строению, выделение из продуктов нефтепереработки- аренов, алкснов, алкадиенов и алкинов ректификацией* как правило, малоэффективно и часто практически невозможна из-за близких температур кипения компонентов и образования азеотропов. Например, бензол образует азеотропы с циклогек-саном, циклогексеном, метилциклопентаном, алканами С7 изо-•строения. При разделении подобных смесей широкое применение находят экстракция, абсорбция, экстрактивная и азеотроп-ная ректификация. Общим для всех этих процессов является использование селективных растворителей, энергия взаимодействия которых с разделяемыми компонентами различна.
Углеводородные системы можно рассматривать в первом приближении как идеальные, подчиняющиеся закону Рауля:
Pi = pQixi> 0)
оде pi — парциальное давление t-ro компонента; Р°{—давление насыщенного пара г-го компонента при данной температуре; Xi — моЛяриая доля /-го компонента в растворе.
В этом случае коэффициент относительной летучести ос разделяемых компонентов рассчитывается как отношение давлений насыщенного пара:
Значения коэффициентов активности углеводородов различных гомологических рядов (прй одинаковом числе углеродных атомов в молекулах) в полярных растворителях изменяются, как правило, в последовательности:
алкаиы > цнклоалкаиы > алкеиы > алкадиены > алкииы > арены.
Именно в¦этой последовательности возрастают силы притяжения между молекулами углеводородов и растворителя.
Алкены, алкины и арены, в отличие от насыщенных углеводородов, являются донорами л-электронов и способны к специфическому взаимодействию — образованию л-комплексов с молекулами электроноакцепторных растворителей С:
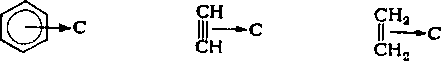
Стабильность я-комплексов возрастает с увеличением элек-тронодонорной способности молекул углеводорода и усилением неравномерности распределения зарядов в молекулах растворителей. В отдельных случаях л-комплексы настолько стабильны, что имеют характерные температуры плавления, например комплексы би- и полицнклических аренов с пикриновой кислотой, 1,3,5-тринитробензолом, 2,4,7-тринйтрофлуореноном,. комплекс бензола с пентафторнитробензолом.
Другой вид специфического взаимодействия углеводородов с полярными растворителями — образование водородных связей, характерное-в особенности для а-алкинов.
В той же последовательности, от алканов к аренам увеличивается и поляризуемость на единицу объема молекул, а следовательно, и энергия вандерваальсова, в частности индукционного, взаимодействия.
Чем больше различаются энергии взаимодействия разделяемых компонентов с молекулами растворителей, тем выше селективность растворителя. Селективность увеличивается при снижении температуры в связи с большей стабильностью я-ком-плексов н при увеличении концентрации растворителя в системе. Максимальная селективность при данной температуре достигается при бесконечном разбавлении углеводородов:
SMaKc = Y?/Y2. (6>
где у 1 н V 2 — коэффициенты активности разделяемых углеводородов при-бесконечном разбавлении растворителем.
Значения 5Макс легко определяются с помощью метода газожидкостной хроматографии, и их удобно использовать для сравнения селективности различных растворителей. В табл.5.2 приведена селективность наиболее эффективных разделяющих агентов, применяемых в процессах экстракции, абсорбции, экстрактивной и азеотропной ректификации, по отношению к системе гексан — бензол. Эта углеводородная система может быть-использована в качестве модельной, так как вследствие единства механизма межмолекулярных взаимодействий растворителей с различными аренами и непредельными углеводородами выполняются достаточно надежно линейные зависимости между значениями селективности при выделении бензола и по отношению к другим аренам с различным числом циклов, а также при выделении алкснов и алкадиенов.
Селективные растворители избирательно растворяют арены или непредельные углеводороды в процессах экстракции и абсорбции, увеличивают коэффициенты относительной летучести насыщенных углеводородов в процессах экстрактивной и азеотропной ректификации.
бензола (ув) и селективность растворителей при 60 °С
|
Растворитель | 0 vr | 0 V6 |
|
| Днэтнленглнколь | 64 | 6,5 |
.9,8 |
|
Трнэтнлеигликоль |
40,5 | 4,2 |
9,6 |
|
Тетраэтиленглнколь (50°С) |
31,8 | 2,58 |
12,3 |
|
Этилеиглнколь | 300 | 20,5 | 15 |
| Сульфолаи |
48 | 2,45 |
19,6 |
|
Днметилсульфокснд |
39 | 3,05 |
12,8 |
|
Днметнлформамид | 11,5 | 1,4 | 8,3 |
| N-Метнлпнрролндон |
8,6 | 1,0 |
8,6 |
|
N-Формнлморфолни (50 °С) |
37,8 | 1,95 |
19,4 |
| Фенол | 12 | 2,5 |
4,8 |
|
Фурфурол | 18 |
2,6 | 6,9 |
|
Метанол | 19 |
5,8 | 3,3 |
|
Ацетон | 5,1 |
1,6 | 3,2 |
|
Ацетоиитрнл | 15,8 |
2,6 | 6,0 |
Азеотропная ректификация с использованием сравнительно низкокипящнх растворителей, таких как метанол, ацетон, ацетонитрил, применяется для выделения аренов (бензола, толуола, ксилолов) из смесей с насыщенными углеводородами, а также для очистки аренов. Азеотропная ректификация с более высококипящими растворителями—бутилцеллозольвом, мо-нометиловым эфиром диэтиленгликоля, диметиловым эфиром тетраэтиленгликоля, триэтиленгликолем использовалась для препаративного выделения аренов Сд — Сю из узкокипящих нефтяных фракций, а такЗке для разделения ареновпроизводных бензола, тетралина и нафталина.
Системы с положительными отклонениями от закона Рауля (к ним, как правило, относятся системы углеводород — полярный растворитель) азеотропны, если выполняется условие:
где индексы 1 и 2 относятся к углеводороду и растворителю соответственно.
Из условия (7) вытекают важные следствия: образование азеотропа тем более вероятно, чем более неидеальна система и чем менее различаются давления насыщенного пара (или температуры кипения) компонентов.
В связи с тем, что полярные растворители образуют более неидеальные системы с насыщенными углеводородами, последние отгоняют в колонне азеотропной ректификации в виде
азеотропных смесей с растворителем, а арены остаются в кубовом остатке.
Азеотропная ректификация находит ограниченное применение при выделении углеводородов вследствие присущих ей недостатков— узкого выбора растворителей, ограниченного условием (7), сравнительно низкой селективности азеотропобразую-щих компонентов и дополнительного расхода теплоты на их испарение. Азеотропная ректификация остается экономически выгодным процессом разделения при очистке целевого продукта от примесей, которые могут быть отогнаны при добавлении небольшого количества азеогропобразующего компонента.
Экстрактивная ректификация отличается использованием сравнительно высококипящих растворителей, которые, как правило, не образуют азеотрспов с разделяемыми компонентами. Для этого температура кипения растворителей должна на 50 °С и более превышать температуры кипения компонентов смеси.
Одно из преимуществ экстрактивной ректификации по сравнению с азеотропной состоит в возможности создания высокой концентрации растворителя в колонне (75—90 %), что повышает селективность и эффективность разделения. При азеотропной же ректификации содержание растворителя в системе определяется составом азеотропов и часто недостаточно велико, что снижает эффективность разделения. Кроме тога, растворители, применяемые при экстрактивной ректификации, характеризуются более высокой селективностью, чем азеотроп-образующие компоненты.
Экстрактивная ректификация редко используется при разделении нефтяных фракций для последующего их анализа, но широко применяется в промышленности для выделения и очистки алкенов, алкадиенов (бутадиена, изопрена), а также для выделения аренов (бензола и его гомологов, стирола) из продуктов пиролиза и каталитического риформинга нефтяных фракций.
Одни и те же селективные растворители, например N-метил-пирролидон или диметилформамид, можно использовать для выделения как аренов, так и алкадиенов. Иногда один и тот же растворитель применяют в различных процессах разделения: так, ацетонитрил успешно используют для выделений 1,3-бутадиена методом экетрактивиой ректификации; он может служить азеотропобразующим компонентом при извлечении аренов из смеси с насыщенными углеводородами.
Важнейшее требование к растворителям для экстрактивной ректификации — сочетание высокой селективности с достаточно большой растворяющей способностью: на тарелках колонны не должно происходить расслаивания жидкой фазы. Этим требованиям удовлетворяют N-метилпирролидон, диме-тилформамид и N-формилморфолин. О высокой растворяющей способности этих растворителей свидетельствуют сравнительно низкие значения коэффициентов активности углеводородов (см. табл. 5.2).
В США процесс экстрактивной ректификации с 1990 г. применяется в промышленности для выделения циклогексана из фракции газоконденсата. При этом используется смесь высокоселективного растворителя с растворителем, повышающим растворяющую способность. Коэффициент относительной летучести ключевой наиболее трудно разделяемой пары 2,3-диметилпен-тан — циклогексан а — 1,22 при массовом отношении растворителя к сырью 7: 1 позволяет выделять циклогексан чистотой 99 % при степени извлечения 90 %.
Для разделения углеводородов можно использовать и процесс экстрактивно-азеотропной ректификации, который проводят в присутствии двух селективных растворителей — азеотроп-образующего компонента и экстрактивного агента. Эти функции может выполнять и один селективный растворитель, т. е. экстрактивный агент в этом случае образует гомогенные или гетерогенные азеотропы с одним из компонентов или группой компонентов разделяемой смеси и попадает как в кубовый остаток, так и в дистиллят.
Экстракция применяется в нефтеперерабатывающей промышленности для выделения аренов из катализатов риформинга бензиновых фракций, а также для селективной очистки смазочных масел от компонентов с низкими индексами вязкости— полициклических ароматических и гетероатомных соединений. Преимущество процесса экстракции состоит в возможности совместного выделения аренов Сб — Се из фракции катализата риформинга 62—140 °С. В процессе же экстрактивной ректификации необходимо предварительное разделение сырья на узкие фракции — бензольную, толуольную и ксилольную с последующим выделением аренов в разных колоннах. Последнее необходимо в связи с тем, что, как вытекает из уравнения (4), коэффициент относительной летучести углеводородов в процессе экстрактивной ректификации зависит не только от коэффициентов активности, но и от давлений насыщенного пара. Поэтому высококипящие насыщенные углеводороды, например С8—Сд, и в присутствии селективного растворителя могут иметь меньшую летучесть, чем бензол, т. е. четкого группового разделения углеводородов не произойдет.
Недостаток экстракции состоит в невысоких значениях коэффициента полезного действия тарелок экстракционных колонн. Экстракционные колонны, роторно-дисковые экстракторы имеют, как правило, эффективность до 10—15 теоретических ступеней, а колонны экстрактивной ректификации — до 100 и более теоретических тарелок. Главным образом по этой причине экстракция не нашла применения для выделения бутадиена и изопрена.
В качестве экстрагентов аренов можно использовать растворители с меньшей растворяющей способностью и, как правило, с большей селективностью — ди-, три- и тетраэтиленгликоль, сульфолан, диметилсульфоксид, смеси N-метилпирролидона или N-метилкапролактама с этиленгликолем.
Для селективной очистки нефтяных масел в качестве экстрагентов применяют фенол, фурфурол, N-метилпирролидон, смеси фенола с крезолами. При производстве остаточных масел проводят предварительную деасфальтизацию гудрона. Для этого компоненты масел экстрагируют неполярными растворителями, например жидким пропаном, и отделяют от асфальтенов.
Экстракцию полярными растворителями можно использовать для разделения моно-, би- и трициклических аренов. Двухступенчатой экстракцией 86- и 91 %-й серной кислотой предложено выделять серосодержащие соединения, в частности сульфиды, из среднедистиллятных нефтяных фракций.
Таким образом, экстракцию применяют и для препаративного разделения, и для анализа нефтяных фракций.
Абсорбция селективными растворителями (N-метилпир-ролидоном, диметилформамидом) используется в промышленности для выделения ацетилена из продуктов окислительного пиролиза природного газа. Ранее для этой цели применяли менее селективные растворители—ацетон, метанол, аммиак, но процесс абсорбции приходилось для повышения селективности проводить при низкой температуре с использованием хлад^ агентов.
Селективные абсорбенты: водные растворы моноэтанол
амина или метилдиэтаноламина, смеси алканоламинов или алифатических аминов с метанолом — используют и при переработке газов для удаления сероводорода, диоксида углерода, серооксида углерода.
Метилдиэтаноламин более селективен при абсорбции сероводорода, чем моноэтаноламин, более стабилен (не взаимодействует с диоксидом углерода), менее коррозионно-агрессивен й по этим причинам получил широкое применение в качествё .абсорбента на зарубежных установках.
Наиболее стабильны амины, в которых аминогруппа при-? соединена к четвертичному атому углерода — 2-амино-2-метшЙ пропанол, терпиндиамин. Такие амнны по образуют карбаматов с диоксидом углерода, что позволяет поддерживать концентрацию свободного амина на высоком уровне, независимо от концентрации С02 в исходном газе.
Наряду с аминоспиртовой очисткой для абсорбции кислых газов в промышленности применяется карбонат калия.
Для отбензинивания нефтяного попутного и природного газов применяют абсорбцию неполярными абсорбентами — углеводородными фракциями. Процесс проводят при температуре окружающей среды или при —40 °С с использованием хладагентов. Преимущество низкотемпературной абсорбции состоит в возможности применения более низкомолекулярных бензиновых фракций с меньшей вязкостью, что повышает эффективность процесса разделения и снижает расход абсорбента.
5.4. КРИСТАЛЛИЗАЦИЯ
И ЭКСТРАКТИВНАЯ КРИСТАЛЛИЗАЦИЯ
Метод кристаллизации применяется для выделения из нефтяных фракций индивидуальных углеводородов или групп углеводородов (например, н-алканов), имеющих наиболее высокие температуры кристаллизации. Температура кристаллизации зависит от размеров молекул и степени их симметрии. Так, температура кристаллизации ^КрИст н-алканов повышается с увеличением их молекулярной массы и, начиная с гептадекана (^крист 22,5 °С), это твердые вещества при комнатной температуре. Температура кристаллизации п-ксилола (13,26°С) на 38,5 °С выше, чем о-ксилола, и на 61 °С выше, чем ж-ксилола. Это объясняется наибольшей степенью симметрии молекул п-ксилола и соответственно наибольшей плотностью их упаковки в кристаллической решетке.
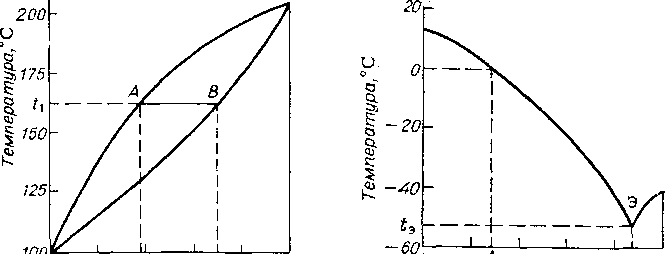
.Фазовые диаграммы равновесия жидкость — твердая фаза могут быть двух типов: с образованием твердого раствора или эвтектической смеси. Фазовая диаграмма первого типа характерна, например, для системы фенантрен — антрацен (рис. 5.1). При охлаждении системы до температуры t\ образуется жидкая фаза А и равновесная твердая фаза В, обогащенная более высокоплавким компонентом—антраценом, однако получение чистого антрацена одноступенчатой кристаллизацией невозможно.
Диаграмма второго типа характерна, например, для системы л-ксилол — лг-ксилол (рис. 5.2). При понижении температуры смеси заданного состава А до 0°С начинается кристаллизация n-ксилола. При дальнейшем снижении температуры вплоть до эвтектической точки t3 (—52,7°С) увеличивается выход твердой фазы, представляющей собой n-ксилол, а состав жидкой фазы изменяется в соответствии с кривой равновесия. При —52,7°С кристаллизуется эвтектическая смесь, и вся система затвердевает, поэтому охлаждение не доводят до эвтектической температуры и кристаллы n-ксилола отделяют фильтрованием или центрифугированием.
inrilc.-1_-U-1-1-1-
0 20 40 60 60 100
-60-Hi—1-1-l—i
0 20A 40 60 80 100
Массовое содержание м-ксилола, %.
Массовое содержание антрацена,%
Рис. 5.1. Фазовая диаграмма для системы фенантрен—антрацен Рис. 5.2. Диаграмма плавкости для системы я-ксилол — ж-ксилол
Присутствие в смеси кроме п- и ж-ксилола других изомеров приводит к снижению температуры кристаллизации эвтектической смеси до —101 °С. При осуществлении процесса в промышленности сырье охлаждают до —(60ч-70)°С.
Получение чистого высокоплавкого компонента одноступенчатой кристаллизацией и для систем с образованием эвтектической смеси практически невозможно: в кристаллах неизбежно остается некоторое количество маточного раствора в результате адсорбции на поверхности, включений в порах и полостях кристаллов, проникновения в трещины под действием капиллярных сил. Поэтому n-ксилол приходится очищать перекристаллизацией или расплавлением части продукта и концентрированием примесей в непрерывных противоточных пульсацион-ных колоннах. Недостатки процесса кристаллизации — низкая степень извлечения n-ксилола (как правило, менее 65% от содержания его в сырье), а также возможность выделения лишь одного, наиболее высокоплавкого компонента.
Кристаллизацией выделяют также дурол (1,2,4,5-тетраме-тилбензол) — наиболее высокоплавкий изомер из алкилбензолов. Сю, возможно выделение нафталина и некоторых алкилнафта-линов, например 2,3,6-триметилнафталина.
Эффективным методом очистки веществ является зонная плавка. Расплавленная зона, образующаяся при нагревании твердого продукта, перемещается между двумя твердыми фазами. Может использоваться и метод зонного вымораживаиия, при котором расплавленный продукт очень медленно застывает.
Аппарат снабжен несколькими обогреваемыми кольцами, между которыми находятся охлаждающие устройства. Вращение трубки с очищаемым веществом позволяет перемешивать жидкую фазу, особенно на поверхности раздела жидкость — твердая фаза, что улучшает теплообмен и повышает скорость прохода зоны.
Экстрактивная кр и ст а л л и з а ц и я — метод кристаллизации с использованием селективных растворителей. Растворитель выполняет несколько функций: селективно растворяет низкоплавкие компоненты; обеспечивает существование жидкой фазы при температуре ниже температуры застывания эвтектической 'смеси, что позволяет повысить выход высокоплавкого компонента; снижает вязкость маточного раствора, что способствует более полному удалению жидкой фазы на стадии фильтрования.
Экстрактивная кристаллизация может использоваться в препаративных целях для выделения высокоплавких циклоалканов, аренов, н-алканов. Так, фракционной кристаллизацией из метанола выделяли адамантан, 1- и 2-метиладамантан из концентратов, полученных перегонкой нефти с водяным паром и последующим комплексообразованием с тиомочевиной.
В нефтеперерабатывающей промышленности экстрактивная кристаллизация получила применение для депарафинизации масляных фракций. Удаление н-алканов, имеющих сравнительно высокую температуру кристаллизации, необходимо для снижения температуры застывания масел и обеспечения их хорошей текучести. Растворитель для этого процесса должен быть достаточно селективным, т. е. должен иметь низкую растворяющую способность по отношению к н-алканам и высокую— к остальным компонентам масляной фракции. Кроме того, растворители должны иметь низкие вязкость и температуру застывания. Наиболее широко в качестве растворителей применяют смеси кетонов (метилэтилкетона, ацетона) с аренами, например толуолом, добавление которого повышает растворимость масляных компонентов и выход масла. С увеличением числа углеродных атомов в молекулах кетонов их селективность снижается, но возрастает растворяющая способность по отношению к масляным компонентам, поэтому, например, метилизобутилкетон можно использовать для депарафинизации масел в индивидуальном состоянии.
На ряде зарубежных установок используется менее селективный растворитель — жидкий пропаи, в этом случае для повышении селективности процесс депарафинизации приходится проводить при более иизкой температуре. В последние годы получила применение смесь пропилеиа с ацетоном, обеспечивающая большую селективность и в связи с этим более низкую температуру застывания масел.
Известна три типа аддуктов и комплексов углеводородов с различными соединениями:
твердые комплексы, образующиеся в результате сильных специфических (электронных донорно-акцепторных) взаимодействий;
аддукты туннельного типа с полостями в кристаллической решетке в виде каналов, в которых находятся молекулы углеводородов или других соединений линейного строения с поперечным сечением, соответствующим диаметру канала;
клатратные соединения с полостями в кристаллической решетке в виде клеток, размеры и формы которых соответствуют молекулам включаемого компонента — «гостя».
Все типы аддуктов и комплексов получили применение для группового разделения нефти и нефтяных фракций.
Молекулярные соединения аренов с сильными электроноакцепторными соединениями. Арены, в особенности полицикли-ческие с конденсированными ароматическими кольцами, являются активными донорами я-электронов и могут образовывать твердые комплексы с сильными электроноакцепторными соединениями.
Давно известны комплексы нафталиновых и других поли-циклоароматических углеводородов с пикриновой кислотой (2,4,6-тринитрофенолом). Пикратным методом из ароматической части фракций 200—300°С был выделен нафталин и ряд его гомологов. Комплексы образуются при нагревании и выделяются вымораживанием, причем обработку ароматической фракции пикриновой кислотой для повышения степени извлечения нафталиновых углеводородов повторяют несколько раз„ пока не начинает вымораживаться чистая пикриновая кислота. После перекристаллизации пикратов их разлагают обработкой эфирных растворов — 2—3%-м водным раствором щелочи.
Для выделения нафталиновых углеводородов из керосиновой фракции в качестве акцепторов можно использовать и другие полинитросоединения, причем стабильность образуемых ими комплексов изменяется в следующем ряду:
2,4,7-тринитрофлуоренон > 1,3,5-трииитробензол > пикриновая кислота >
> 2,4,6-тринитротолуол.
Комплексообразованием с n-нитробензойной кислотой можно выделять 2,6-диметилнафталин из технической смеси диметил-нафталинов, а с 3,5-динитробензойной кислотой — мезитилен из смеси аренов С9.
Стабильные комплексы с нафталином и его алкилпроизвод-ными (в особенности с осевой симметрией молекул) с темпера-
турами плавления ~ 150 -f- 230°С дает пиромеллитовый диангидрид.
Ряд гомологов бензола образует достаточно стабильные комплексы с гексафторбензолом. Предложен, например, метод разделения изомеров ксилола кристаллизацией в присутствии гексафторбензола, образующего наиболее стабильные комплексы с ж-ксилолом.
Один из промышленных методов выделения л-ксилола из смеси аренов С8 основан на комплексообразовании с борофтороводородной кислотой BF3HF, возможно использование для этой цели и трифторметансульфокислоты F3CSO3H.
Сильным электроноакцепторным соединением является и пентафторнитробензол, образующий твердый комплекс с бензолом.
Для образования комплексов с аренами могут использоваться и неорганические акцепторы — хлорид или бромид алюминия, хлорид сурьмы(Ш).
Комплексообразование углеводородов с карбамидом и тио-карбамидом. Комплексы углеводородов с карбамидом (мочевиной) и тиокарбамидом (тиомочевиной) относятся к аддуктам туннельного типа.
В 1940 г. немецкий исследователь М. Ф. Бенген установил, что алифатические соединения с линейной структурой молекул, в частности н-алканы, содержащие шесть и более атомов углерода, образуют кристаллические комплексы с карбамидом. Разветвленные алканы, циклоалканы и арены, как правило, ие способны к комплексообразованию с карбамидом.
Карбамид — сильно ассоциированное соединение из-за образования межмолекулярных водородных связей:
NH2 Н О
NH2—С=0 . . . Н—N—С—NH2.
Как показали рентгеноструктурные исследования, карбамид имеет тетрагональную кристаллическую решетку, которая при образовании комплекса изменяется на гексагональную. Структура комплекса характеризуется расположением ассоциированных молекул карбамида по спирали на гранях правильных шестигранных призм (рис. 5.3). Элементарная ячейка состоит из шести молекул карбамида, находящихся на расстоянии 0,37 нм друг от друга. Внутри спирали образуется канал гексагональной формы эффективным диаметром 0,525 нм. Поперечное сечение молекул н-алканов составляет около 0,42 нм, поэтому они хорошо вписываются в канал и удерживаются в нем за счет сил Ван-дер-Ваальса. Молекулы разветвленных алканов, циклоалканов и аренов имеют критические диаметры, превышающие эффективный диаметр канала, и поэтому, как правило, не способны образовать аддукты с карбамидом.
Рис. 5.3. Схема кристаллической решетки комплекса карбамида:
•о — атомы кислорода молекул карбамида; • — атомы кислорода одной элементарной
ячейки
Стабильность комплексов возрастает с удлинением цепи н-алкана. Так, температура разложения комплекса к-гексана с карбамидом 38°С, энтальпия разложения комплекса 21 кДж/моль, а для н-додекана— соответственно 90,9°С и 54 кДж/моль. Образование аддукта с пентаном и н-алканами с С < 5 энергетически невыгодно, и при комнатной температуре и атмосферном давлении выделить соответствующие комплексы не удается.
Аддукты с карбамидом способны образовывать не только н-алканы, но и углеводороды других классов, молекулы которых имеют достаточно длинный алкильный неразветвленный заместитель. Так, если метальная группа находится в положении 2,3,4 или 5 молекулы монометил алкана, то для возможности образования аддукта в линейном участке цепи должно содержаться не менее соответственно 11, 14, 15 или ,16 углеродных атомов. Циклические углеводороды также способны к образованию комплексов, если боковая цепь линейного строения содержит не менее 18 углеродных атомов.
По этой причине селективность выделения н-алканов карба-мидным методом снижается с повышением пределов кипения нефтяной фракции, и наиболее эффективна карбамидная депа-рафинизация сырья с концом кипения ^350°С. Концентрат, выделенный из более высококипящей фракции карбамидным методом, например из фракции 350—500°С вакуумного газойля западносибирской нефти, содержит всего 73 % н-алканов, ¦остальное приходится на долю изоалканов (около 11 %), циклоалканов (14%) и аренов (2%).
Комплексы н-алканов с карбамидом относятся к нестехиометрическим соединениям включения — соотношение между числом молей компонентов в комплексе нецелочисленное. Так, молярное соотношение карбамид/н-алкан составляет для гексана 5,43, для додекана 9,42 и т. д. Предложены различные эмпирические формулы для расчета этого соотношения, из которых
следует, что на каждую СНг-группу молекулы н-алкана должно приходиться около 0,7 моль карбамида.
Комплексы с тиокарбамидом NH2C(S)NH2 также относятся' к соединениям включения туннельного типа. Водородные связи с участием атома серы менее стабильны, чем в случае карбамида, расстояние между молекулами тиокарбамида соответственно увеличивается и образуется канал с большим диаметром (0,6—0,7 нм по данным различных авторов). Поэтому в качестве молекул «гостя» могут выступать алканы изостроения, циклоалканы, некоторые арены. К ним относятся углеводороды изопреноидного строения, циклогексан, декалин, ада-мантан, дурол. н-Алканы, как правило, не дают стабильных аддуктов с тиокарбамидом, так как поперечное сечение их молекул значительно меньше диаметра канала и сравнительно слабые вандерваальсовы силы притяжения не способны удерживать н-алканы внутри канала.
Тиокарбамид — менее селективный разделяющий агент, чем карбамид. Тиокарбамидцый метод позволяет лишь концентрировать определенные группы углеводородов в препаративноаналитических целях. Так, в сочетании с другими методами комплексообразование с тиокарбамидом позволяет получать, концентраты алканов изопреноидного строения, адамантана и его гомологов, разделять цис- и гранс-диалкилциклогексаны: с очень близкими температурами кипения.
Комплексообразованием с тиокарбамидом в присутствии активатора (метанола) предложено выделять метилциклопен-тан и циклогексан.
Соединения включения туннельного типа способны образовывать с углеводородами и ряд других веществ — дезоксихоле-вая кислота (диаметр канала 0,5—0,6 нм), 4,4'-дигидрокситри-фенилметан (канал 0,6—0,65 нм), 7-циклодекстрин (канал 0,9—1,0 нм). Однако эти соединения не получили столь широкого применения в практике разделения нефтей, как карбамид или тиокарбамид.
Клатратные соединения с полостями в кристаллической решетке в виде клеток. Клатратные соединения впервые открыты Г. Дэви, установившим в 1811 г., что хлор с водой образует-твердый газовый гидрат. Несколько позже были' проведены первые исследования и гидратов углеводородов—метана, этана,, этилена, пропана.
В 1886 г. Ф. Милью обнаружил, что гидрохинон образует комплексы с инертными газами: азотом, аргоном, ксеноном, криптоном. Химических связей в этом случае образоваться не могло, поэтому оставалось предположить, что комплексы формируются в результате полного окружения молекул инертного-газа молекулами гидрохинона.
Впоследствии это предположение было подтверждено рентгенографическими исследованиями: ассоциированные за счет водородных связей молекулы гидрохинона образуют трехмерный каркас, включающий молекулы второго компонента. Г. М. Пауэлл предложил называть подобные соединения клат-ратами— от латинского clathratus, что значит «включенный» или «заключенный за решетку».
Молекулы «гостя» могут быть связаны в клатрат, если их размеры и форма соответствуют геометрическим размерам ячеек в кристаллической решетке молекул «хозяина». На этом •основано разделение углеводородов, в частности изомеров ксилола.
Для разделения изомеров ксилола использовали комплексы Вернера, имеющие общую формулу: металл-(лиганд)4-(анион)2. В качестве металла чаще всего используют никель, лигандами •служат азотсодержащие основания, например 4-метилпиридин или бензиламин. Так, используя тетракис(4-метилпиридин)дироданид никеля
![]()
можно выделить n-ксилол из смеси ксилолов. При одноступенчатом выделении n-ксилола его объемное содержание повышается с 19,3 до 64%. Этот же комплекс Вернера селективен и по отношению к другим пара-изомерам, в частности к п-этил-толуолу.
При замене лиганда — использовании вместо 4-метилпири-дина 4,4'-дипиридила — или при замене тиоцианатного аниона формиатным можно селективно выделять о-ксилол. Этилбензол образует клатратные соединения с тетракис(4-ацетилпиридин)-дироданидом никеля и выделяется из смеси аренов Се-
Осаждение клатратов проводят при пониженной температуре. Метод применяли в промышленных масштабах в США для выделения n-ксилола, однако он не получил широкого распространения из-за больших расходов комплекса Вернера (около 5 ч. на 1 ч. n-ксилола), коррозии оборудования, токсичности солей никеля и производных пиридина.
Клатратные соединения — газовые гидраты — образует вода с низшими алканами, некоторыми серосодержащими соединениями, а также циклопентаном и циклогексаном.
Газовые гидраты — это нестехиометрические соединения включения, имеющие общую формулу М-пНгО, где М—молекула гидратообразователя, а п ^ 5,67. По внешнему виду это твердые кристаллические вещества, напоминающие снег или рыхлый лед. Однако кристаллическая решетка газовых гидратов отличается от кристаллической решетки льда стабильностью при температуре выше 0°С и наличием внутренних полостей определенных размеров, доступных для молекул ряда соединений, в частности для метана, этана, пропана, изобутана, этилена, пропилена, ацетилена.
Структура газовых гидратов была установлена в результате исследований М. Штакельберга в 40—50-х годах. В присутствии гидратообразователей может образоваться кристаллическая решетка двух различных типов из молекул воды, связанных между собой водородными связями. Элементарная ячейка структуры первого типа состоит из 46 молекул воды и содержит две малые полости в форме додекаэдров со средним диаметром 0,52 нм и 6 больших полостей — тетрадекаэдров со средним диаметром 0,59 нм. Элементарная ячейка структуры второго типа состоит из 136 молекул воды и содержит 16 малых (диаметр 0,48 нм) и 8 больших полостей (диаметр 0,69 нм). Если максимальный размер молекул «гостя» меньше 0,48 нм,, то в кристаллической структуре второго типа могут оказаться заполненными все полости — как большие, так и малые. При этом п в общей формуле газовых гидратов принимает минимальное значение, равное 5,67.
Метан и углеводороды С2 образуют газовые гидраты со-структурой первого типа, а изобутан и пропилен гидраты состава М-17Н20, что соответствует заполнению только больших полостей структуры второго типа. Бутан, и высшие гомологи с максимальным размером молекул больше 0,69 нм не участвуют в процессе гидратообразования.
Для повышения стабильности гидратов можно использовать вспомогательный газ — сероводород, молекулы которого заполняют малые полости кристаллической решетки, тем самым стабилизируя ее. Молекулы углеводородов включаются в большие полости.
Стабильность гидратов определяется не значением критического диаметра молекул углеводорода, как это имеет место при комплексообразовании с карбамидом или адсорбции на цеолитах, а зависит от максимального размера молекул «гостя»-(табл. 5.3).
Алканы с температурами кипения, близкими к температуре кипения циклопентана и циклогексана, например гексан, длина молекулы которого (1,03 нм) больше размера клеток в кристаллической решетке гидратов, не способны к образованию" водных клатратов даже в присутствии вспомогательного газа. Поэтому, проводя клатратообразование при 0—18°С с 0,4—
0,7 %-м водным раствором сероводорода, можно выделять цик-логексаи и циклопентан, например, из газоконденсатной и изо-меризатной фракции.
Предложено использовать газовые гидраты для опреснения морской воды. Например, жидкий пропан при перемешивании
Таблица 5.3. Температура разложения водных клатратов циклических углеводородов (вспомогательный газ — сероводород)
| Углеводород |
Максимальный размер молекул, нм | Температура разложения (при 0,1 МПа), °С |
|
Циклопентаи | 0,56 |
19,8 |
|
Циклопентен | 0,58 |
17,2 |
|
Циклогексан | 0,60 |
15,3 |
|
Цнклогексен | 0,62 |
10,0 |
|
1,3-Циклогексаднен |
0,66 | 9,3 |
| Бензол | 0,69 ' |
6,5 |
с морской водой образует гидраты, а растворенные в воде соли в гидратную решетку не проникают. Другое возможное применение газовых гидратов состоит в хранении в виде гидратов природных, а также инертных газов.
Интересны исследования по разделению бинарных и многокомпонентных смесей с помощью водных клатратов, в частности до демеркаптанизации природного газа.
Образованием гидратов, забивающих трубопроводы и аппаратуру, может сопровождаться ряд процессов в нефтедобывающей, газовой и нефтехимической промышленности. Для предотвращения возникновения гидратов и разрушения уже образовавшихся пробок можно использовать следующие методы: повышение температуры (подогрев газа горячей водой или паром); снижение давления; снижение содержания воды в газе путем осушки, вымораживания или применения специальных добавок (гликолей, спиртов), снижающих парциальное давление паров воды.
5.6. АДСОРБЦИЯ
Выделение некоторых классов соединений, присутствующих в нефтях и нефтепродуктах, осуществляется с большей избирательностью на адсорбентах, чем с помощью селективных растворителей.
Алкены несколько лучше растворяются в селективных растворителях, чем алканы с тем же числом углеродных атомов, что создает принципиальную возможность их разделения экстракцией. Однако растворимость углеводородов в полярных растворителях снижается н гомологических рядах с увеличением молекулярной массы. Поэтому н смесях широкого фракционного состава растворимости алкенов и алканов взаимно перекрываются и разделить их экстракцией практически невозможно. Использование же адсорбционного метода позволяет решать эту задачу.
Различают адсорбенты с неупорядоченной кристаллической структурой и неоднородной пористостью (силикагель, активный оксид алюминия, активные угли) и адсорбенты с однородными порами — цеолиты или молекулярные сита.
Разделение нефтяных фракций на адсорбентах с неоднородной пористостью. Наиболее широкое применение среди этой группы адсорбентов получили силикагели, что объясняется возможностью варьирования в широких пределах их адсорбционных характеристик, негорючестью, относительной дешевизной. Силикагель — это высушенный гель кремниевой кислоты. В России его выпускают в гранулированном и кусковом виде. В зависимости от пористой структуры силикагели подразделяют на мелкопористые и крупнопористые, которые классифицируют маркам в зависимости от размеров зерен.
Выпускают следующие марки отечественных силикагелей: КСМГ — крупный силикагель мелкопористый гранулированный,. KCKJ — крупный крупнопористый, ШСМГ и ШСКХ — шихта-силикагель мелко- и крупнопористый, АСКГ—активированный (неудачное название более мелкой фракции, остающейся при получении крупного силикагеля) и кусковые силикагели КСМК, ШСМК, МСМК и АСМК (последняя буква шифра «К» — кусковой). Размер зерен крупного силикагеля 2,8—7,0 мм, шихты 1,0—3,6 мм, мелкого силикагеля 0,25—2,0 мм и активированного 0,2—0,5 мм.
В крупнопористых силикагелях содержится 94 % SiC>2 и в качестве примесей А!203 (0,2—0,5 %), Fe203 (до 0,1%), оксиды щелочных и щелочноземельных металлов. Мелкопористые силикагели в качестве упрочняющей добавки содержат 7—10% А120з, содержание SiC>2 в них около 89%. Средний диаметр пор мелкопористых силикагелей составляет 1,7—2,8 нм в зависимости от марки, а для крупнопористых силикагелей 14,9—17,2 нм. Наилучшими адсорбционными характеристиками отличается силикагель КСМК — У него наиболее развитая удельная поверхность (~760 м2/г) и мелкие поры (1,7 нм), размеры которых изменяются в сравнительно узком интервале значений.
Выбор марки силикагеля зависит от размера молекул адсорбируемых компонентов. Например, для разделения и анализа керосино-газойлевых и масляных фракций используют крупнопористые силикагели (АСКГ), для осушки углеводородов — мелкопористые силикагели.
Адсорбируемость на полярных адсорбентах, к которым относятся силикагели, тем выше, чем больше дипольный момент или диэлектрическая постоянная вещества. Активные центры поверхности силикагеля наиболее сильно специфически взаимодействуют с гетероатомными компонентами нефтяных фракций. Хорошо сорбируются на силикагелях также полициклические арены, несколько слабее—арены с одним ароматическим кольцом и значительно слабее — алканы и циклоалканы.
К полярным адсорбентам относится и оксид алюминия. Использование оксида алюминия позволяет более четко разделять арены на моно-, би- и полициклические и несколько лучше отделять углеводороды от сероорганических соединений. Оксид алюминия можно использовать и для селективного выделения алкинов из смесей с алканами.
Неполярные адсорбенты—активные угли — неспецифически взаимодействуют с разделяемыми компонентами. Их можно использовать для анализа газовой смеси, а также для разделения жидких алканов, изоалканов и циклоалканов. Однако селективность разделения невысока, так как активные угли характеризуются наиболее неоднородной пористостью — диаметр пор от 2 до нескольких сот нанометров.
Разделение нефтей и нефтепродуктов с помощью цеолитов. Цеолиты — наиболее селективные адсорбенты, обладающие упорядоченной кристаллической структурой и определенным размером входных окон. Поэтому цеолиты, называемые также молекулярными ситами, способны сорбировать только те молекулы, критический диаметр которых меньше эффективного диаметра окон.
Название «цеолит», в переводе с греческого, означающее «кипящий камень», было дано еще в XVIII веке в связи со способностью природных цеолитов вспучиваться при нагревании в результате выделения из пор воды. Природные цеолиты встречаются как в изверженных, так и в осадочных породах. В настоящее время установлены структуры и химический состав 34 видов природных цеолитов. Наиболее распространены клиноптилолит, имеющий следующий состав: (Na2K2)OX
X Al203-10Si02-8H20 и содержащийся в туфах Закавказья, а также морденит и филлипсит. Избирательная адсорбция веществ с критическим диаметром молекул не более 0,5 мм была установлена в 1925 г. для одного из природных цеолитов — шабазита.
В конце 50-х годов были разработаны удобные методы производства синтетических цеолитов нагреванием водных щелочных алюмосиликатных смесей. Цеолиты имеют следующий состав:
М2/„0 • А1203 • xSi02 • «/Н20,
где М — щелочной илн щелочноземельный металл (Na, К, Mg, Са, Ва, Sr), л — валентность металла.
В промышленности выпускают цеолиты различных структурных типов: к(х = 2), Х(л; = 2,4 ч- 2,8) и высококремнеземные цеолиты типа Y(x = 4,8).
В настоящее время синтезированы еще более высококремнеземные цеолиты с соотношением Si02/Al203 до 10:1, наибольшее содержание кремния характерно для синтетического мордеиита. Синтезировано более 100 видов цеолитов, в том числе около 25 идентичных природным. В качестве адсорбентов и катализаторов используют главным образом синтетические цеолиты, они эффективнее и высококачественнее природных.
Каркасная структура цеолита включает тетраэдры SiO* и АЮ4, образующие основную ячейку решетки — кубооктаэдр. Замещение Si4+ на Als+ приводит к остаточному отрицательному заряду решетки, который нейтрализуется катионами щелочных или щелочноземельных металлов, расположенными во внутренних полостях решетки.
В цеолитах типа А ионы Si4h и А13+ соседних кубооктаэдров связаны кислородными мостиками из четырех ионов кислорода, а в цеолитах типа X — из шести ионов кислорода. Малые поры —объемы внутри кубооктаэдрических структурных единиц— одинаковы для цеолитов типа А и X. Большие поры (полости)— пространства между кубооктаэдрами и кислородными мостиками — также незначительно различаются для разных типов цеолитов по размерам. Основные различия состоят в размерах окон больших полостей. Большие полости цеолитов типа А сообщаются между собой восьмичленными кислородными окнами, а окна цеолитов типа X включают 12 ионов кислорода и отличаются большим диаметром. Диаметр окон зависит также от природы катиона.
В соответствии с классификацией цеолитов, принятой в России, указывается катион, преимущественно входящий в решетку, и тип кристаллической решетки. В марках США и ряда других зарубежных стран указывается диаметр входных окон (в А) и тип решетки. Данные различных исследователей об эффективном диаметре окон цеолитов типа X существенно расходятся.
Ниже приводится эффективный диаметр d окон цеолитов различных марок (для цеолитов типа X — по данным М. М. Дубинина) :
| Россия | США |
d, нм |
| КА | ЗА | 0,3 |
| NaA | 4А | 0,4 |
|
СаА | 6А |
0,6 |
| СаХ |
ЮХ | 0,8 |
|
NaX | 13Х |
0,9 |
Изоалканы с одной метильной груп- 0,65 пой в боковой цепн
Алканы с двумя метальными груп- 0,67 памн
|
водородов, нм: | |
|
Метан | 0,40 |
| я-Алканы С3—Си |
0,49 |
| Бензол | 0,57 |
|
Циклогексан | 0,61 |
Алканы с одной этнльной группой 0,72
В соответствии с критическими размерами молекул и диаметром окон цеолит КА адсорбирует практически только воду; NaA—воду, СОг, H2S, ЫНз,.СНзОН, этилен, пропилен, низшие алкадиены и алкины, • этан; СаА—нормальные углеводороды и спирты с числом углеродных атомов до 20, метил- и этилтио-спирты, этиленоксид. Цеолит СаХ адсорбирует разветвленные алканы и спирты, бензол, циклогексан и их низшие гомологи. На СаХ не сорбируются соединения ароматического характера с разветвленными радикалами или большой молекулярной массой, например 1,3,5-триэтилбензол, 1,3-дихлорбензол.
Цеолиты—полярные адсорбенты, поэтому адсорбционное разделение веществ на них можно проводить, используя не только разницу в молекулярных размерах, но и различную степень ненасыщенное™ и полярности. Критический диаметр-сильно адсорбируемых полярных молекул углеводородов с двойными и тройными связями может даже несколько превышать диаметр окон.
Цеолиты типа NaY имеют следующий состав:
Na20 • AI2O3 • 4,8Si02 • 8,9Н20,
т. е. в них содержится вдвое меньше тетраэдров АЮ4 и соответственно катионов по сравнению с цеолитом.типа X. Размеры входных окон цеолитов X и Y одинаковы. Цеолиты типа Y отличаются повышенной кислотостойкостью.
Разделение углеводородов и нефтяных фракций на цеолитах широко применяется как в препаративно-аналитических целях, так и в промышленности. Адсорбцией на цеолите СаА из керо-сино-газойлевых фракций (200—320°С) выделяют н-алканы Сю — С1 s, которые используют для микробиологического синтеза белков, а также для производства биологически разлагаемых моющих веществ. Адсорбция проводится, как правило,, в паровой фазе, так как в случае жидкофазного процесса трудно с достаточной полнотой отделить несорбируемые компоненты от слоя сорбента. При десорбции н-алканов в качестве вытеснителей используют аммиак, пентан, гексан.
Успешно используют и комбинированные методы разделения, в которых адсорбционная депарафииизация сочетается с процессами каталитического риформинга, изомеризации и ал-килирования. В этих процессах адсорбцией н-алканов на цеолите СаА удается повысить октановое число бензина на 13—21.
Адсорбция на цеолитах применяется и для выделения не-разветвленных алкенов Сю — Cie из смесей с алканами.
Адсорбционное разделение ксилолов на цеолитах более эффективно и экономично, чем кристаллизация и экстракция. Степень извлечения n-ксилола при одноступенчатом адсорбционном процессе составляет 98,4%, при кристаллизации — 60%, а при экстракции — 80%. Лучше всего для адсорбции п-ксилола использовать цеолит ВаУ или цеолиты типа X и Y в бариевой и калиевой формах при массовом отношении Ва: К от
5 до 35. Цеолит типа RbY селективен по отношению к этилбен-золу, CaY и SrY — к .«-ксилолу, a NaY — к о-ксилолу.
Цеолиты — прекрасные осушители газов и жидкостей, а также хорошие поглотители серосодержащих соединений.
Природные и модифицированные кислотой клиноптилолит-содержащие туфы — активные и селективные адсорбенты гете-роатомных соединений нефти. Наиболее полно выделяются тиоспирты — степень демеркаптанизации реактивных топлив достигает 94—97 %, при этом значительно повышается термостабильность и другие показатели качества топлив. Для удаления гетероатомных соединений и аренов из реактивных топлив можно использовать и синтетические цеолиты NaX и СаХ. При адсорбционном разделении на цеолите CaNaX деароматизиро-ванных керосиновых фракций сорбируются преимущественно би- и трициклические циклоалканы, отличающиеся высокой теплотой сгорания — важным показателем для реактивных топлив.
Цеолиты используют и при анализе нефтяных фракций в качестве неподвижной фазы в газовой адсорбционной хроматографии.
5.7. ТЕРМИЧЕСКАЯ ДИФФУЗИЯ
Сущность явления термодиффузии, открытого К. Людвигом в 1856 г., состоит в том, что при наличии температурного градиента в смеси, состоящей из нескольких компонентов, возникает градиент концентраций. Заполнив U-образную трубку раствором сульфата натрия и поддерживая в одном ее колене температуру 0, а в другом 100°С, Людвиг обнаружил через некоторое время в холодном колене выпавшие в осадок кристаллы соли.
После изобретения термодиффузионной колонки (1938 г.) термическую диффузию стали использовать для разделения смесей, трудноразделимых другими методами, в том числе нефтяных фракций. Термодиффузионные колонки состоят из двух коаксиальных цилиндров с зазором между ними 0,25—0,5 мм. Разделяемую смесь помещают в пространство между цилиндрами, один из которых нагревают, а другой охлаждают. При этом молекулы одного вещества перемещаются к холодной стенке или цилиндру и в результате конвекции опускаются вниз, а молекулы другого компонента направляются к горячему цилиндру и концентрируются в верхней части колонки. Основные закономерности процесса: 1) к холодной стенке движется углеводород с наибольшим числом углеродных атомов и с наибольшей температурой кипения; 2) при одинаковой температуре кипения к холодной стенке направляется компонент с наименьшим молярным объемом; 3) при одинаковых молярных объемах и температурах кипения к холодной стенке движется компонент с наименьшей поверхностью молекул.
Как правило, термодиффузионному разделению подвергают сравнительно узкокипящие (25—50-градусные) фракции, предварительно разделенные на алкан-циклоалкановую и ареновую части. При термической диффузии насыщенных углеводородов
б верхней части колонки концентрируются алканы, в средних фракциях — моно- и бициклоалканы и в последних термодиффузионных фракциях (в нижней части колонки) — полицикло-алканы. Таким образом, метод термодиффузии позволяет более или менее успешно решать следующие сложные задачи:
разделять смеси близкокипящих циклоалканов и алканов изостроения (например, можно разделять циклогексан и 2,4-ди-метилпентан, температуры кипения которых различаются всего на 0,24°С);
разделять смеси циклоалканов по числу циклов с получением концентратов моно-, би- и полициклоалканов;
разделять цис-, транс-изомеры, например цис- и транс-декалин, цис- и транс- 1,2-диметилциклогексан.
Степень разделения компонентов повышается, если навить проволоку по винтовой линии на внутреннюю трубку колонки — при этом уменьшается влияние паразитной конвекции. К тому же эффекту приводит использование роторных диффузионных колонн, в которых внутренний цилиндр медленно вращается. Недостаток как роторных колонн, так и колонн со спиральной навивкой состоит в увеличении продолжительности достижения стационарного состояния, которое составляет, как правило, десятки часов.
Невысокая производительность термодиффузиоиных колонн ограничивает возможности использования метода в промышленности. Тем не менее, предлагалось использовать термодиффузию для получения фракций масел с высоким индексом вязкости и низкой температурой застывания. Имеются зарубежные установки производительностью до 9 т/сут смазочного масла с повышенным индексом вязкости.
Разработаны автоматизированные аппараты непрерывного термодиффузионного разделения (АТР-3 и АТР-ЗМ), которые успешно используют для препаративного разделения нефтяных фракций и нефтехимических продуктов.
5.8. ДИФФУЗИЯ ЧЕРЕЗ МЕМБРАНЫ
Разделение газов и жидких смесей углеводородов диффузией через непорист'ые полимерные мембраны основано на различии в форме молекул разделяемых компонентов и их растворимости в материале мембраны.
Перенос вещества через непористые мембраны включает стадии сорбции, диффузии и десорбции с противоположной стороны мембраны. Обычно сорбция и десорбция протекают быстро по сравнению с диффузией, скорость которой определяет суммарную скорость переноса. Скорость переноса в соответствии с первым законом Фика выражается следующим образом:
/ = ?> (C,-C,)/L.
где D—коэффициент диффузии; L — толщина мембраны; Ct и С2—концентрации компонента на граничных поверхностях мембраны.
Если разделяется газовая смесь и выполняется закон Генри: С == ар, то
j = Do (р2 — pO/L.
Таким образом, скорость переноса зависит как от условий эксперимента (L, Ар), так и от свойств системы полимер — пенетрант (D, о). Величина P—Da выражается в см3-см/(см2Х Хс-Па) и называется коэффициентом проницаемости. Она характеризует объем газа в см3, приведенный к нормальным условиям, проходящий в 1 с через 1 см2 мембраны толщиной 1 см при перепаде парциального давления 1 Па. Высокой проницаемостью должны обладать вещества с малым поперечным сечением молекул (с высокими значениями D), хорошо растворимые в материале мембраны (с большими коэффициентами растворимости а).
В табл. 5.4 приведены значения D, а и Р для различных газов и паров в поливинилтриметилсилане, используемом для мембранных процессов газоразделения.
Как следует из табл. 5.4, высокие коэффициенты проницаемости водорода являются следствием малых размеров его молекул, а для воды и диоксида углерода — следствием больших коэффициентов растворимости.
Селективность, или фактор разделения,
ati==P{/Pj
определяет степень обогащения бинарной смеси компонентом i при однократном прохождении через мембрану.
и проницаемости пенетрантов в поливинилтриметилсилане при 25°С
|
Пенетрант | Размер | D ¦ 10', | а • 10s, | Р-10'2, |
| молекулы, нм |
см2/с |
см5/(см3 • Па) |
см3 ¦ см/(см2. с- Па) |
|
| н2 |
2,9 | 180 |
0,09 | 16,2 |
| сн4 | 3,3 |
1,0 | 1,06 |
1,0 |
| со2 | 4,0 | М |
2,7 | 13,5 |
| Н20 | 4,2 | 1,2 | 56 |
67,2 |
|
С3Н4 | 4,0 | 0,19 | 21 |
4,0 |
| СзНа | 4,3 | 0,001 |
30 | 0,3 |
Эффективные мембраны должны обеспечивать высокую селективность а,/ и производительность (Pi), иметь хорошую устойчивость в средах.
Основное препятствие для широкого применения мембран — трудность создания мембран с высокой селективностью и с большой газовой проницаемостью. Эти два свойства находятся для большинства мембран в обратной зависимости. Обычно используют высокоселективные полимеры с низкой проницаемостью в виде очень тонких слоев («0,1 мкм) на пористой неселективной полимерной подложке.
Применяются и мембраны с облегченным переносом — пористые, пропитанные селективным растворителем, связывающим один из компонентов, что облегчает его перенос через мембрану.
На зарубежных промышленных установках реализованы мембранные процессы концентрирования водорода из водородсодержащих газов установок гидроочистки, гидрокрекинга и риформинга. В качестве мембран используют полые волокна из полисульфона или поливинилтриметилсилана. Применяют также процессы разделения смесей Н2/СО для корректировки состава синтез-газа в процессах оксосинтеза. С помощью мембран из ацетилцеллюлозы в виде спирально изогнутых элементов, помещенных в трубки Пито, выделяют H2S и СОг из метансодержащего газа, проводят осушку газа. Реализовано в промышленности и мембранное концентрирование кислорода и азота из воздуха.
Имеются принципиальные решения и перспективы мембранного разделения смесей метан — этилен, этилен — пропан, ал-лен — пропан, бензол — циклогексан и некоторых других. Дальнейший прогресс мембранных процессов разделения зависит от синтеза полимеров для более высокоселективных и стабильных мембран.
Химические методы разделения и идентификации компонентов нефти и газа в значительной степени утратили свое значение с развитием хроматографии и других физических и физикохимических методов. Однако в ряде специфических случаев химические методы необходимы для полного разделения нефти, особенно для выделения гетероатомных соединений и непредельных углеводородов.
Химические методы разделения основаны на различной реакционной способности компонентов в реакциях гидрирования, дегидрирования, сульфирования, изомеризации, галогенирова-ния и т. д. Так, реакция каталитического гидрирования имеет аналитическое значение для гетероатомных соединений, которые переводят таким образом в сравнительно легко анализируемые углеводороды. Комбинирование реакции дегидрирования циклоалканов до аренов со скелетной изомеризацией пятичленных циклоалканов, которая протекает с расширением цикла, позволило дать полную характеристику различных типов циклоалканов в нефтяных фракциях.
Очистка изопрена и бутадиена от алкинов может быть осуществлена селективным гидрированием последних. Для удаления непредельных и ароматических углеводородов из смесей с насыщенными углеводородами можно использовать реакцию, сульфирования.
На различии в скоростях сульфирования и гидролиза образующихся сульфокислот основан также метод разделения изомеров ксилола и этилбензол.а. Скорость сульфирования м-ксилола больше, чем других изомеров, вследствие согласованного ориентирующего влияния метальных групп в его молекуле. Скорость гидролиза ж-ксилолсульфокислоты также наибольшая. Применяя ступенчатое сульфирование и ступенчатый гидролиз образующихся сульфокислот (постепенно повышая температуру и концентрацию серной кислоты), можно выделить сначала ж-ксилол, а затем последовательно разделить остальные изомеры.
Для выделения алкадиенов из смесей с алкенами и насыщенными углеводородами можно использовать хемосорбциои-ные методы, основанные на образовании комплексов с различной стабильностью между непредельными углеводородами и солями металлов переходной валентности, в частности солями меди(1) и серебра.
Интересны также микробиологические методы разделения углеводородов, в частности депарафинизация газойлевых фракций. Микроорганизмы используют в качестве питательной среды м-алкаиы, в результате получаются синтетический белок и депарафинизироваиный газойль. Предложен также микробиологический метод обессеривания нефти. Под действием некоторых микроорганизмов серосодержащие соединения превращаются в водорастворимые продукты, легко удаляемые из нефти.
5.10. МЕТОДЫ ВЫДЕЛЕНИЯ ОТДЕЛЬНЫХ ГРУПП УГЛЕВОДОРОДОВ
Алканы. Выделение н-алканов — относительно несложная задача, которая решается следующими методами.
Ректификация — для разделения смесей сравнительно низкокипящих алканов, например н-бутана и изобутана, н-пен-тана и изопентана.
Адсорбция на цеолитах типа СаА — самый селективный метод выделения н-алканов, применяющийся как в промышленности, так и в аналитической практике. В качестве сырья могут использоваться прямогонные бензиновые и керо-сино-газойлевые фракции с концом кипения не выше 350°С.
Карбамидная депарафинизация — для удаления н-алканов из керосино-газойлевых и масляных фракций, при этом получают низкозастывающие дизельные топлива зимних сортов и трансформаторные масла.
Желательно, чтобы в сырье для процесса карбамидной де-парафинизации не было смол и серосодержащих соединений, так как они являются ингибиторами комплексообразования. Поэтому, как правило, проводят предварительную гидроочистку сырья. Кроме того, при производстве алканов желательно их высокое содержание в исходной фракции (>20%) и низкое содержание аренов (не выше 15%).
Большинство вариантов процесса карбамидной депарафини-зации предусматривает введение активаторов — веществ, ускоряющих процесс комплексообразования. В качестве активаторов предложены и применяются спирты (наиболее эффективен метанол), кетоны, нитроалканы. Активаторы препятствуют адсорбции ингибиторов на кристаллах карбамида. Кроме того, активаторы, растворяя часть карбамида, способствуют протеканию процесса в гомогенной среде с большей скоростью.
К исходной смеси добавляют еще и растворитель для снижения вязкости, обеспечения тесного контакта карбамида с н-ал-канами, облегчения транспортирования образующихся суспензий. Проведение процесса с растворителем повышает чистоту выделяемых н-алканов. Применяемые растворители можно подразделить на три группы: 1) растворяющие углеводороды и плохо растворяющие карбамид — бензин, толуол, некоторые спирты, кетоны; 2) растворяющие карбамид и плохо растворяющие углеводороды — вода, водные растворы низших спиртов; 3) растворяющие как углеводороды, так и карбамид — изопропиловый и изобутиловый спирты, метилизобутилкетон и др. Некоторые растворители (ацетон, метилэтилкетон, изопропиловый спирт, метиленхлорид, смеси нитроалканов) могут служить одновременно и активаторами.
Карбамидную депарафинизацию проводят при 20—35°С. Повышение температуры увеличивает взаимную растворимость веществ, снижает их вязкость и улучшает условия контакта, однако стабильность комплексов и выход н-алканов при этом уменьшаются. Выбор температуры зависит от требуемой степени депарафинизации, пределов кипения исходной фракции и от того, вводится ли карбамид в кристаллическом состоянии или в виде водного раствора. Депарафинизацию сравнительно низкокипящих фракций, а также процесс с использованием водных растворов карбамида для повышения стабильности комплексов необходимо проводить при более низкой температуре.
Недавно разработан процесс извлечения парафинов ком-плексообразованием с карбамидом непосредственно из высоко-парафинистых и парафинистых нефтей.
Экстрактивная кристаллизация — для выделения твердых парафинов из масляных фракций. В качестве селективных растворителей используют смеси метилэтилкетон — толуол, метилэтилкетон — метилизобутилкетон, дихлорэтан — дихлорме-тан, ацетон — пропилен и пропан. Те же растворители можно применять и для обезмасливания парафина.
Микробиологическая депара ф и н изаци я и каталитическая гидродепарафинизация — для удаления н-алканов из нефтяных фракций.
Алканы изостроения выделить из нефтяных фракций значительно сложнее, чем н-алканы. Особенно большие трудности возникают при отделении их от циклоалканов. Близость силовых полей молекул изоалканов и циклоалканов, их неспособность к специфическим взаимодействиям не позволяет использовать для разделения селективные растворители. Критические диаметры молекул также, как правило, приблизительно одинаковы, что приводит к низкой селективности методов, основанных на различии размеров и формы молекул.
Тем не менее, получать фракции, обогащенные изоалканами, можно с использованием следующих методов:
ректификация при различных давлениях применительно к низкокипящим бензиновым фракциям;
комплексообразование с тиокарбамидом — этим методом были сконцентрированы, в частности, изопреноид-иые углеводороды из фракции 200—500 °С. Как известно, ад-дукты с тиокарбамидом образуют молекулы с поперечным сечением 0,68 ± 0,03 нм на 0,58 ± 0,05 нм, что примерно соответствует критическим размерам изопреноидных углеводородов;
термическая диффузия — изоалканы концентрируются в верхней части термодиффузионной колонки.
Циклоалканы. Для концентрирования циклоалканов и разделения их на фракции по числу циклов в молекулах могут быть использованы те же методы, которые применяют для выделения алканов нзостроения. Отделение же циклоалканов от аренов и гетероатомных соединений осуществляется сравнительно легко с помощью селективных растворителей или адсорбционной жидкостной хроматографии.
Выделение низкокипящих циклоалканов (циклопентана, циклогексана) возможно гидратообразованием.
Арены. Выделение аренов из смесей с насыщенными углеводородами проводят в основном с помощью селективных растворителей методами экстракции и экстрактивной ректификации, а при исследованиях состава нефтяных фракций — методом адсорбционной жидкостной хроматографии.
В табл. 5.5 приведены основные условия проведения процесса экстракции аренов (бензола, толуола, ксилолов) из катализаторов риформинга экстрагентами, применяющимися в промышленности.
В 50—60-х годах основным экстрагентом аренов служил днэтиленгликоль — растворитель, достаточно селективный, термически и гидролитически стабильный, сравнительно дешевый и малотоксичный. Основной недостаток диэтиленгликоля — низкая растворяющая способность по отношению к аренам и, как следствие, необходимость проведения экстракции при высоком соотношении экстрагент: сырье и высокой температуре, что приводит к высоким энергетическим затратам.
Таблица 5.5. Оптимальные условия процессов экстракции аренов экстрагентами, применяющимися в промышленности
| Массовое | ||
| Экстрагент |
соотношение экстрагент: сырье | Температура, °С |
| Днэтиленгликоль (с 7 % воды) | 16:1 | 155-160 |
| Триэтиленгликоль (с 5 % воды) | (10ч-11): 1 | 150 |
| Тетраэтиленгликоль (с 5 % воды) | 8:1 | 145—150 |
| Сульфолан (с 1—2 % воды) | 90—100 | |
|
N-Метилпирролидон — этиленгликоль |
(4 -s- 6): 1 | 50—60 |
| (60 : 40) |
6,2:1 | 60—70 |
| N-Метнл-е-капролактам — этнлеиглн- | ||
| коль (30 : 70) |
3,5:1 (об.) (Б+ 6): Г | |
| N-Формилморфолнн | — |
|
| Днметилсульфоксид (с 10 % воды) | 25—35 |
На ряде установок диэтиленгликоль заменен на экстрагент с более высокой растворяющей способностью — триэтиленгли-коль, а за рубежом используется и тетраэтиленгликоль, что. позволяет существенно снизить массовое отношение экстрагента к сырью.
В конце 50-х годов был разработан более эффективный процесс экстракции аренов сульфоланом — растворителем с более высокой как селективностью, так и растворяющей способностью. Степень извлечения аренов из фракции 62—140°С сульфоланом составляет для бензола — 99,9; толуола — 99,5 и ксилолов — 98%. В настоящее время сульфолан является наиболее широко применяемым за рубежом экстрагентом, используется он и на комплексах по производству аренов, в России.
На ряде зарубежных установок применяется смесь N-метил-пирролидона с этиленгликолем, а в Германии — смесь N-метил-е-капролактама с этиленгликолем. Лактамы проявляют очень высокую растворяющую способность по отношению к аренам, а добавление этиленгликоля приводит к повышению селективности и критической температуры растворения сырья.
Высокоселективный растворитель для выделения аренов в основном методом экстрактивной ректификации — N-формил-морфолин.
Французский институт нефти в 60-е годы разработал процесс экстракции ареиов диметилсульфоксидом. Недостаток этого экстрагента — невысокая термическая и гидролитическая стабильность. Поэтому его регенерацию производят не обычным способом ректификации с водяным паром, а реэкстракцией аренов из экстрактной фазы низкокипящими алканами (бутаном, пентаном). Подобную реэкстракционную схему можно использовать в процессах экстракции аренов экстрагентами с очень высокой температурой кипения, например тетраэтилен-гликолем, в качестве реэкстрагента применяют высококипящий алкан (додекан).
Полициклические арены, содержащиеся в масляных фракциях, удаляют экстракцией фенолом, фурфуролом, N-метилпир-ролидоном, нитробензолом, смесями фенола с крезолами. Наиболее широко используемые для этой цели растворители — фенол и фурфурол — имеют существенные недостатки. Фенол малоселективен, что приводит к невысокому качеству и выходу рафинатов, кроме того, он вызывает ожоги при попадании на кожу, имеет сравнительно высокую температуру кристаллизации. Основной недостаток фурфурола — низкая термоокислительная стабильность, что приводит к большим потерям экстрагента и забивке экстракционного оборудования образующимися полимерами. Нитробензол находит ограниченное применение из-за высокой токсичности и сравнительно высокой температуры кипения, что осложняет регенерацию экстрагента. Наиболее эффективным экстрагентом для селективной очистки масел является N-метилпирролидон.
Для разделения аренов, в частности изомеров ксилола и этилбензола, кроме ректификации (для выделения о-ксилола и на некоторых установках этилбензола) в промышленности применяют адсорбцию на цеолитах, кристаллизацию «-ксилола, экстракцию .м-ксилола борофтороводородной кислотой.
При исследованиях состава нефтяных фракций арены нафталинового ряда можно выделять комплексообразованием с пикриновой кислотой или другими сильными электроноакцепторными соединениями.
Углеводороды фенантренового ряда выделяют из нефтяных фракций, используя реакцию фотоконденсации с малеиновым ангидридом:
соон
ноос
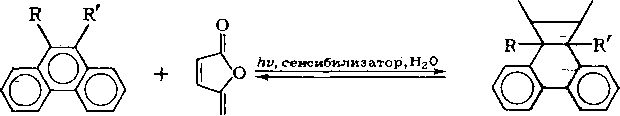
о
Наличие заместителей в положениях 9 и 10 приводит к увеличению выхода аддуктов, однако если эти заместители имеют большой объем, то вследствие пространственных препятствий выход продукта реакции снижается. Антрацен и его гомологи взаимодействуют с малеиновым ангидридом даже в темноте: -
о
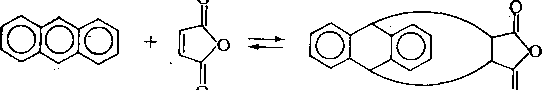
о
о
Производные бензола и нафталина не реагируют с малеиновым ангидридом. Поэтому сначала проводят обработку фракции малеиновым ангидридом в темноте и выделяют углеводороды антраценового ряда, а затем на свету, и выделяют фен-антрен и некоторые его гомологи. Однако выход аддуктов производных фенантрена с малеиновым ангидридом составляет всего около 40 %.
Алкены и алкадиены. Алкены и алкадиены, получаемые при пиролизе нефтяных фракций или дегидрированием алканов, содержат также алканы н другие примеси. Разделение продуктов пиролиза состоит из следующих основных стадий.
1. Компрессия газа пиролиза и выделение из него углеводородов С5 и выше. Компримированием газа до 3,5—4 МПа в многоступенчатых компрессорах и межступенчатой сепарацией газы пиролиза отделяют от основной массы конденсирующихся высших углеводородов и воды.
2. Очистка газа пиролиза от Н2, С02 и органических серосодержащих соединений. Газы пиролиза очищают от сероводорода абсорбцией водным раствором моноэтаноламина (или ме-тилдиэтаноламина), протекающей с образованием органических солей:
20-40° с
2HOCH2CH2NH2 + H.S--->(Hnr.H?r.H,NH,U s.
100-110° с
При содержании серы в сырье пиролиза менее ОД % можно ограничиться для очистки газа как от Н2, так н от С02 промывкой водным раствором щелочи. При этом частично удаляется серооксид углерода:
2NaOH + COS —> NaHC03 + NaHS.
3. Осушка газов пиролиза путем абсорбции воды диэтилен-гликолем и затем на цеолите NaA.
4. Удаление ацетилена и его производных, а также аллена СН2=С = СН2 селективным гидрированием в присутствии палладиевого или никелькобальтхромового катализатора. Эту стадию часто проводят после предварительного выделения этан-этиленовой и пропан-пропиленовой фракций.
На крупнотоннажных пиролизных установках целесообразно выделять ацетилен, метилацетилен иаллен. Так, на установках ЭП-300 образуется до 6 тыс. т/год ацетилена и суммарно до 5 тыс. т/год аллена и метилацетилена. Разработаны процессы выделения этих ценных мономеров экстрактивной ректификацией с ацетонитрилом или диметилформамидом.
5. Выделение фракций С2, Сз, С4 и получение концентрированных алкенов путем ректификации. Примерные условия газо-разделения и средние коэффициенты относительной летучести аср ключевых пар компонентов приведены в табл. 5.6.
Для выделения водорода и метана из очищенного газа пиролиза используют низкотемпературную ректификацию под давлением. Коэффициент относительной летучести ключевой пары компонентов метан — этилен, как следует из табл. 5.6, достаточно высок, поэтому метановая колонна имеет около 30 тарелок.
Перспективны, как отмечалось выше, и уже начинают применяться для выделения водорода и низкокипящих углеводородов процессы диффузии через полимерные мембраны.
Деэтанизация — выделение этан-этиленовой фракции (ключевые компоненты — этан и пропилен) осуществляется сравнительно легко ректификацией в колоннах, имеющих около 40 тарелок.
Т а б л и ц а 5.6. Средние значения коэффициентов
относительной летучести пар компонентов
| Ключевые компоненты | р, МПа |
t, | °с | “ср | |
| легкий | тяжелый | в конденсаторе | в кипятильнике | ||
| сн4 |
С2Н4 | 3,43 | —92,8 | -7,8 | 5,3 |
| С2Н4 | С2Нв |
2,06 | —27,8 | -5,6 |
1,48 |
| С2Нв | CsHs | 2,06 |
—5,6 | 55,6 | 3,0 |
| СзНв | СзНв |
1,54 | 37,8 | 43,3 | 1,15 |
|
с2н8 |
ИЗО-С4Н10 | 1,37 | 37,8 | 82,2 | 2,06 |
| С2Н10 | H30-CsHi2 |
0,36 | 37,8 | 71,1 | 2,06 |
Более сложная задача — выделение этилена из этан-этиле-новой фракции. Ректификацию, проводят в колоннах со 110— 120 тарелками при флегмовом числе 4,5—5,5. Еще более эффективные колонны, содержащие до 200 тарелок, требуются для выделения чистого пропилена при флегмовом числе около 10.
Предложен ряд других методов выделения пропилена: экстрактивная ректификация, адсорбция на цеолитах NaA, силикагеле и алюмогеле, хемосорбция растворами солей меди(1) в полярных растворителях. Возможна также микробиологическая очистка пропилена: пропускают пропан-пропиленовую
фракцию и воздух через микробиологический раствор, пропан служит питательной средой для микроорганизмов, а пропилен получается очищенным. Однако эти методы выделения пропилена не получили промышленного развития и наиболее экономичным процессом остается ректификация.
Молярное содержание в пиролизной фракции изобутилена около 27% и 1,3-бутадиена свыше 35%. Выделить эти углеводороды ректификацией не удается из-за близких температур кипения компонентов и образования азеотропных смесей.
Методы выделения изобутилена основаны на его повышенной реакционной способности по сравнению с н-бутиленами, которая объясняется повышенной электронной плотностью двойной связи из-за эффекта сверхсопряжения шести С—Н-свя-зей с двойной связью. Так, скорость абсорбции изобутилена 60 %-й H2SO4 в 150—200 раз выше, чем 2-бутенов, и приблизительно в 300 раз выше, чем 1-бутена. Недостатки этого метода— наличие высокоагрессивных коррозионных сред, относительно низкая чистота продукта, высокий выход олигом'еров.
В меньшей степени эти недостатки присущи модификации процесса с 40—50 %-й H2SO4. При этом происходит гидратация изобутилена в трет-бутиловый спирт, который выделяют ректификацией и дегидратируют:
h,so4
(СН3)2С = СНа + Н20 (СНз)зСОН.
В России применяется также метод гидратации изобутилена на ионитных катализаторах.
Разработан метод выделения изобутилена, основанный на взаимодействии с соляной кислотой в присутствии хлоридов металлов:
S21CI2
(СНз)2С = СН2 + НС1 ^ (СНз)з СС1.
Образуется смесь трет- бутилхлорида и грег-бутилового спирта, которые отделяют, и при нагревании до 85—120°С выделяют чистый изобутилен.
При взаимодействии фракции С4, содержащей изобутилен, с уксусной кислотой образуется трет-бутилацетат, который отделяют и при нагревании получают изобутилен.
Цеолиты с диаметром входных окон 0,8 нм из фракции С4 адсорбируют только изобутилен, что может быть использовано для его выделения и очистки.
Наиболее перспективный процесс выделения грег-алкенов — превращение их при взаимодействии со спиртами в простые алкил-трет-алкиловые эфиры. Так, изобутилен взаимодействует с метанолом с образованием метил-трет-бутилового эфира — высокооктанового компонента автобензинов. В качестве катализаторов используют сульфокатионит, формованные иойиты. Катализаторами на стадии получения изобутилена разложением эфира могут служить оксид алюминия, алюмосиликаты, фосфорная кислота на кизельгуре, ионообменные смолы. До обработки спиртом из фракции С4 должны быть выделены диеновые углеводороды.
Разработан также метод разделения изобутилена и н-буте-нов на основе изомеризации 1-бутена в сравнительно высоко-кипящий 2-бутен с последующим выделением более летучего изобутилена ректификацией. Катализаторами процесса служат сульфокатиониты, палладий на оксиде алюминия.
Выделяют 1,3-бутадиен из пиролизной фракции С4, а также из продуктов дегидрирования бутана в основном экстрактивной ректификацией. В табл. 5.7 приведены коэффициенты относительной летучести углеводородов С4 по отношению к 1,3-бутадиену без растворителя и в присутствии применяющихся селективных растворителей.
Выделение 1,3-бутадиена из фракции С4 экстрактивной ректификацией проводится по двухстадийной схеме. В первой колонне отгоняются насыщенные углеводороды и бутены, массовое соотношение растворитель : сырье при использовании ацетонитрила 4,2:1, эффективность колонны — 70 теоретических тарелок (т. т.). Во второй колонне (40 т. т.) при массовом соотношении ацетонитрила к сырью 2,3:1 отгоняется 1,3-бута-днен от алкинов.
Таблица 5.7. Коэффициенты относительной летучести углеводородов С4 без растворителя и при массовом содержании растворителей 70 % (температура 50 °С)
|
Без | Диметил- |
N-Мети л- | ||
|
Углеводород | растворителя | Ацетоиитрил |
формамид | пирроли дои |
|
Изобутан | 1,165 |
3,38 | 3,35 |
3,34 |
| я-Бутан | 0,85 | 2,42 |
2,40 | 2,38 |
| Изобутилеи |
1,03 | 1 >75 |
1,78 | 1,75 |
| 1 -Бутеи | 1,03 | 1,75 | 1,78 |
1,75 |
|
граме-2-Бутен | 0,83 | 1,43 | 1,46 |
1,46 |
|
цис-2-Бутен | 0,764 | 1,30 | 1,30 |
1,30 |
|
1,3-Бутадиеи | 1,00 |
1,00 | 1,00 | 1,00 |
| 1,2-Бутадиен |
0,625 | 0,725 |
0,70 | 0,70 |
| 2-Бутин | 0,378 |
0,33 | 0,33 |
0,35 |
| 1-Бутин | 0,67 | 0,50 |
0,47 | 0,48 |
| Бутении | 0,72 | 0,42 | 0,31 |
0,31 |
Технико-экономические показатели процессов с ацетонитрилом, диметилформамидом и N-метилпирролидоном практически одинаковы. Преимущество ацетонитрила состоит в меньшей температуре кипения и, соответственно, меньшей термополимеризации бутадиена.
Очистку 1,3-бутадиена проводят ректификацией в двух колоннах.
Ранее для выделения бутадиена использовали процесс хемосорбции, основанный на способности алкенов образовывать координационные соединения с солями металлов переменной¦ валентности, например с водно-аммиачным раствором ацетата меди(1):
fHsNv /Ov 1 ГС^Нвх /0\ 1
;с< ^ССНз +2С4Нв ;С< ;ССНз +2NH3.
Lh3n/ \о/ J Lc4He/ \о/ J
Растворимость углеводородов в хемосорбенте уменьшается в следующем ряду: 1,3-бутадиен > 1-бутен > цыс-2-бутен >
> изобутилен > транс-2-бутен. Растворимость 1,3-бутадиена, отличающегося более высокой электронодонорной способностью по сравнению с алкенами, приблизительно в 10 раз выше растворимости 1-бутена. Процесс хемосорбции проводят при «—10°С, а десорбцию бутадиена из хемоэкстракта — при нагревании до 80—90 °С.
Выделение и очистку изопрена из пиролизной фракции С5 продуктов дегидрирования изопентана проводят методом экстрактивной ректификации с диметилформамидом или N-ме-тилпирролидоном. В табл. 5.8 приведены коэффициенты относительной летучести углеводородов фракции С5 без растворителя и в присутствии диметилформамида.
Выделение изопрена проводят в двух колоннах экстрактивной ректификацией. При использовании диметилформамида в первой колонне эффективностью 50—55 т. т. при соотношении растворитель : сырье 8: 1 отгоняются насыщенные углеводороды и алкены (ключевая пара компонентов — 2-метил-2-бу-тен — изопрен). Во второй колонне (90 т. т.) при соотношении растворитель: сырье 4:1 отгоняется изопрен (чистота 99,3%), а в кубовом остатке остаются пиперилены (пентадиены), циклопентадиен, алкины.
Затем простой ректификацией из изопрена удаляют примеси более летучих алкинов (2-бутин, 3-метил-1-бутин). Выделенный изопрен дополнительно очищают, так как требования к его чистоте для стереорегулярной полимеризации очень жесткие. Допустимое содержание примесей в изопрене в 10—20 раз ниже, чем в бутадиене для производства бутадиенового каучука. Сильными каталитическими ядами при полимеризации изопрена являются циклопентадиен, 1-алкины, карбонильные, серо- и азотсодержащие соединения — их допустимое содержание исчисляется десятитысячными долями процента.
Первоначальную очистку изопрена от менее летучих примесей— пипериленов и большей части циклопентадиена — осуще-
Таблица 5.8. Коэффициенты относительной
летучести углеводородов фракции Cs без растворителя и в присутствии диметилформамида при массовом содержании 75 % и температуре 50° С
|
Углеводород | Вез |
Диметилформ- |
|
растворителя | амид | |
| Изопентан |
1,209 | 3,02 |
| м-Пентаи | 0,926 | 2,39 |
|
Циклопентаи | 0,61 |
1,14 |
|
3-Метил-1-бутен | 1,535 | 2,63 |
|
2-Метил-2-бутен | 0,874 | 1,39 |
|
Г-Пентен | 1,136 |
1,90 |
|
гра«с-2-Пеитеи |
0,909 | 1,55 |
| Чые-2-Пентен | 0,913 | 1,54 |
|
Циклопеитен | 0,719 | 0,91 |
|
Изопрен | 1,00 |
1,00 |
|
транс-1,3-Пентадиен |
0,783 | 0,76 |
| Ч«с-1,3-Пеитадиен |
0,726 | 0,70 |
| Циклонентадиен |
0,811 | 0,62 |
| 2-Пентин | 0,484 |
0,44 |
|
3-Метил-1-бутин | 1,318 | 0,89 |
|
1-Пентин | 0,842 |
0,55 |
| 2-Метил-1 -бутсн-3-ин | 1,05 1,29 | 0,49 |
| 2-Бутин | 0,90 |
ствляют ректификацией. Затем проводят тонкую очистку от циклопентадиена, основанную на использовании реакции Тиле:
0+о-< - 0=<+н-°-
При взаимодействии циклопентадиена с карбонильными соединениями (циклопентаноном, ацетоном, бензальдегидом, аце-тофеноном, метилэтилкетоном и др.) образуются высококипя-щие фульвены, от которых изопрен легко отделяют ректификацией. Кислородсодержащие соединения удаляют промывкой водой.
Очистка изопрена от 1-алкинов достигается каталитическим гидрированием. По конверсии при гидрировании с никелевым катализатором на кизельгуре алкины располагаются в ряд:
1-пентин > 2-метил-1-бутен-З-ин > 3-метил- 1-бутин > 3,3-ди-метил-1-бутин >- 2-бутин > 2-пентин. Изопрен гидрируется в значительно меньшей степени: при остаточном массовом содержании 1-алкинов 0,0002 % конверсия изопрена составляет 1-2%.
Заключительные стадии очистки изопрена — азеотропная осушка и очистка от низкокипящих примесей (в основном от
2-бутина), а также высококипящих соединений — осуществляются ректификацией.
В пиролизной фракции С5 содержится 12—15 % пипериле-нов (цис- и транс-1,3-пентадиены), которые также мЬгут быть выделены экстрактивной ректификацией с диметилформамидом или другими селективными растворителями.
Из фракции С8 жидких продуктов пиролиза выделяют стирол. Предварительно фенилацетилен, отделение которого простой и экстрактивной ректификацией недостаточно эффективно, избирательно гидрируют. Затем экстрактивной ректификацией с N-метилпирролидоном или диметилформамидом из фракции С8 в первой колонне отгоняют этилбензол и ксилолы, летучесть
Таблица 5.9. Коэффициенты относительной летучести углеводородов фракции С8 пиролиза без растворители и в присутствии N-метилпирролидоиа при его массовом содержании 50 % и температуре 80° С
| Углеводород |
Без растворителя |
N-метил- иирролпдон |
Углеводород | Вез растворителя | N-метил- пирролидон |
|
Октан 1,95 7,84 Этилбензол 1,^8 1,90 /г-Ксилол 1,29 1,82 ж-Ксилол 1,24 1,78 |
о-Ксилол 1,04 1,46 Кумол 0,80 1,25 Стирол 1,00 1,00 Фснилацетилен 1,04 0,74 |
||||
которых возрастает в присутствии селективных растворителей (табл. 5.9), и во второй колонне выделяют стирол.
При пиролизе бензиновой фракции на 100 тыс. т этилена образуется 4—6 тыс. т стирола. Процесс выделения стирола из продуктов пиролиза впервые реализован в промышленности в Японии.
