Особенности технологии и аппаратурного оформления термических и термокаталитических процессов глава i , особенности технологии деструктивных процессов переработки нефтяного сырья
ОСОБЕННОСТИ ТЕХНОЛОГИИ И АППАРАТУРНОГО ОФОРМЛЕНИЯ ТЕРМИЧЕСКИХ И ТЕРМОКАТАЛИТИЧЕСКИХ ПРОЦЕССОВ
ГЛАВА I ,
ОСОБЕННОСТИ ТЕХНОЛОГИИ ДЕСТРУКТИВНЫХ ПРОЦЕССОВ ПЕРЕРАБОТКИ НЕФТЯНОГО СЫРЬЯ
Для технологии процессов химической переработки нефтяного сырья характерно преимущественное применение высоких температур и зачастую высоких давлений.
Область температур термических процессов — от 500 до 1000 °С. Использование катализаторов позволяет вести процесс при более умеренных температурах, однако в некоторых случаях каталитическим процессам тоже свойственны высокие температуры: каталитический крекинг на цеолитсодержащих катализаторах ведут при 500—540 °С (но при очень коротком времени контакта сырья с катализатором), каталитический пиролиз осуществляют при 650°С (вместо 750—850°С для термического процесса).
Повышенное давление присуше всем процессам химической переработки нефтяного сырья, осуществляемым в присутствии водорода. Так, каталитический риформинг бензинов и гидроочистку светлых продуктов проводят при 1,0 — 5,0 МПа, гидрокрекинг — при давлениях, достигающих 15,0 — 20,0 МПа. При этом парциальная доля водорода в газовой фазе достигает 90% (мольн.), т. е. процесс идет по существу в атмосфере водорода. Высокое давление используют и в некоторых термических процессах с целью повышения пропускной способности установок.
Тепловые эффекты
Для большинства рассматриваемых процессов характерно наличие теплового эффекта суммарных реакций. Этот тепловой эффект может быть отрицательным (и в этом случае для осуществления процесса необходимо затрачивать некоторое количество тепла) или положительным (когда происходит выделение тепла, и для сохранения изотермичности процесса необходимо отводить тепло из реакционной зоны)251.
Реакции разложения, дегидрирования и деполимеризации, сопровождающиеся образованием из исходной молекулы сырья двух и более молекул продуктов разложения, имеют, как правило, отрицательный тепловой эффект, т. е. требуют затрат тепла. Реакции присоединения водорода, полимеризации и конденсации, сопровождающиеся образованием из двух и более молекул одной молекулы большей молекулярной массы, протекают с выделением тепла. Отрицательный тепловой эффект реакций разложения свидетельствует о том, что им благоприятствуют высокие температуры; глубина экзотермических реакций возрастает с понижением температуры. Таким образом, чем селективнее протекает процесс, тем выше его суммарный тепловой эффект, на который в данном случае не влияют другие реакции, протекающие параллельно и обладающие иногда тепловым эффектом, противоположным по знаку.
Например, теплота дегидрирования метилциклогексана в толуол составляет примерно —2219 кДж/кг (—530 ккал/кг), а теплота дегидрирования диметил-циклогексана в ксилол — примерно —1862 кДж/кг (—445 ккал/кг), т. е. в среднем— 2040 кДж/кг (—490 ккал/кг). При каталитическом риформинге бензинов на катализаторе средней активности происходит почти полное превращение иаф-теиов в ароматические углеводороды при относительно малой реакционной способности парафинов, поэтому тепловой эффект процесса в основном определяется содержанием в сырье (в бензине) нафтенов. По данным Маслянского, тепловой эффект каталитического риформинга бензина из восточной иефти, содержащего 22% нафтенов, составляет—251 кДж на 1 кг сырья, а для аналогичной фракции бензина ильской нефти, содержащей 40% нафтенов, он равен —440 кДж/кг. При сопоставлении этих цифр с приведенными выше для чистых нафтеновых углеводородов можно заключить, что если бы протекало только дегидрирование нафтенов, тепловой эффект был бы гораздо ниже:
в первом случае: 2040-0,22=450 кДж/кг
во втором случае: 2040-0,40 = 816 кДж/кг
Пониженные значения теплового эффекта свидетельствуют о параллельном протекании экзотермических реакций, частично компенсирующих затраты тепла на процесс. Действительно, в процессе каталитического риформинга идут и реакции гидрокрекинга, сопровождающиеся выделением тепла.
При проектировании реакционных аппаратов крекинг-установок необходимо знать теплоту реакции. Эту теплоту можно определить экспериментально — посредством калориметра, однако этот метод сложен и не всегда технически осуществим. Иногда теплоту реакции крекинга определяют при помощи закона Гесса.
Согласно закону Гесса, тепловой эффект химической реакции не зависит от пути перехода одного вещества в другое, а зависит только от начального и конечного состояния вещества. Так, если углерод сгорает полностью, то количество выделившегося тепла будет одинаково, независимо от того, произойдет ли сгорание по уравнению
С + 02 —> C02 + Q
или последовательно по двум уравнениям;
С + 0,502 -> СО + Q' СО + 0,502
Отсюда итоговое тепло сгорания равно:
Q = Q'+Q”
При использовании закона Гесса необходимо учитывать агрегатное состояние исходных и образующихся веществ. Естественно, например, что образование водяного пара или воды из водорода и кислорода будет сопровождаться тепловыми эффектами, различающимися на величину, равную скрытой теплоте испарения воды.
Применительно к процессу крекинга нефтяного сырья закон Гесса можно использовать с помощью экспериментальных данных по теплотам сгорания исходного сырья и продуктов крекинга. Если в результате крекинга получены газ, бензин, промежуточная фракция и крекинг-остаток, то, обозначив теплоту сгорания этих продуктов и сырья через Q с соответствующим индексом, получим:
Q — Qr + Сб + Оаф + Qko — Qc
Недостаток метода заключается в том, что вследствие больших значений теплоты сгорания (пятизначные числа) небольшая относительная ошибка в ее определении вызывает значительную ошибку в абсолютных единицах и, следовательно, ошибку в теплоте реакции, порядок цифр которой гораздо меньше. Ошибка может оказаться весьма значительной, если отклонения при определении теплот сгорания сырья и какого-нибудь из продуктов крекинга окажутся с разными знаками (если, например, эти отклонения примерно одинаковы по величине, но разные по знаку, ошибка в определении теплового эффекта процесса будет вдвое больше).
Если взамен экспериментально определенных теплот сгорания пользоваться эмпирическими формулами, то подсчеты по уравнению Гесса абсолютно ненадежны. Более точные результаты можно получить, используя в уравнении Гесса не теплоты сгорания, а теплоты образования сырья и продуктов реакции. Теплоты образования значительно меньше, чем теплоты сгорания. Так, теплота сгорания метана равна «55680 кДж/кг, а теплота его образования всего 4677 кДж/кг.
При определении теплот образования сложных смесей известного группового химического состава можно условно принять, что эти смеси состоят из индивидуальных углеводородов соответствующих температур кипения и молекулярной массы.
Из других способов определения теплового эффекта процессов химического превращения нефтяного сырья следует остановиться на составлении тепловых балансов промышленных реакторов. Если известен материальный баланс реактора и его точные режимные данные, можно, составив тепловой баланс аппарата, определить тепловой эффект по алгебраической разности между приходом и расходом тепла. Для получения более точных результатов необходимо учитывать потери тепла в окружающую среду.
/
Наличие тепловых эффектов требует соответствующего конструктивного оформления реактора. При осуществлении термического или каталитического крекинга, риформинга и других процессов, сопровождающихся затратой тепла на реакцию, необходимо вносить тепло в реакционную зону. Это достигается либо подводом тепла через стенку труб нагревательно-реакционного змеевика печи, либо некоторым перегревом исходного сырья, либо применением твердого или газообразного теплоносителя. В процессах, протекающих .с выделением тепла, для поддержания постоянной температуры необходим отвод тепла; с этой целью применяют прямой ввод охлаждающего агента в реактор или создают там режим, способствующий теплоотводу (через теплоотводящую поверхность). Например, в реакторы гидрокрекинга во избежание подъема температуры вводят холодный водород, а при алкилиро-вании изобутана газообразными олефинами выделяющееся тепло отводят путем испарения части изобутана, находящегося в системе. Конкретные схемы реакционных устройств рассмотрены при описании соответствующих процессов.
Рециркуляция непревращенного сырья
Большая часть химических превращений нефтяного сырья характеризуется протеканием побочных реакций. При этом с углублением процесса роль побочных реакций усиливается и соответственно падает выход целевого продукта.
Глубину процесса обычно характеризуют долей превращенного сырья. Если оно представляет собой индивидуальный углеводород, то количество непревращенного сырья легко определить существующими методами анализа. Применительно к промышленному сырью сложного углеводородного состава понятие глубины превращения, как правило, условно. Так, при крекинге (термическом и каталитическом) за непревращенное сырье обычно принимают ту часть жидких продуктов, которая выкипает в пределах выкипания исходного сырья (например, фракция, выкипающая выше 350°С). Однако такое допущение условно, так как по химическому составу продукт крекинга значительно отличается от сырья, и сходство ограничивается только пределами выкипания. Это различие будет тем больше, чем более глубоко прошел процесс. Назовем поэтому такую фракцию «•условно непревращенной» частью сырья.
В некоторых случаях понятие глубины превращения становится еще более условным. Например, при пиролизе бензина с целью получения газообразных олефинов выход газа доходит до 80%, считая на бензин; при этом наряду с газообразованием происходят столь глубокая ароматизация и уплотнение молекул, что состав жидкого продукта пиролиза (смола) совершенно отличен от состава исходного бензина. При пиролизе за глубину превращения принимают выход газа, но с одинаковым основанием можно принять и выход целевого продукта — этилена.
В ряде случаев процесс превращения сырья целесообразно проводить с определенной, заранее заданной глубиной, возвращая «условно непревращенную» часть сырья в зону реакции. Очевидно, что если состав этой возвращаемой фракции близок к составу сырья, то фракцию перед подачей в систему целесообразно смешать с соответствующей порцией свежего сырья.
Если первичное превращение идет глубоко, то «условно непревращенную» часть сырья можно либо отдельно подвергать вторичному превращению, либо вообще выводить из системы в качестве побочного продукта.
При каталитическом крекинге вакуумного газойля на современных цеолитсодержащих катализаторах образуется тяжелый газойль — высокоароматизированный продукт, склонный к коксо-образованию; его подвергают крекингу в отдельном реакторе или выводят из системы (полностью или частично). Смолу пиролиза никогда не возвращают на повторный процесс, так как она представляет собой смесь ароматических углеводородов с непредельными и еще более склонна к коксообразованию, чем тяжелый газойль каталитического крекинга. При термическом крекинге нефтяных фракций для увеличения выхода бензина целесообразно направлять на повторный крекинг (большей частью в смеси со свежим сырьем) промежуточные газойлевые фракции, которые относительно легко подвергаются крекингу.
Еще более очевидна целесообразность возврата непревращенного сырья в реактор, если сырьем являются индивидуальные углеводороды или их простейшие смеси. Например, при каталитической изомеризации н-пентана с целью получения изопентана (высокооктановый компонент бензинов) допустимая глубина превращения соответствует выходу изопентана 50—65%; при этом образуется очень незначительное количество газообразных продуктов разложения (1—2% на сырье), а остальную часть жидкого продукта (непревращенный н-пентан) возвращают в зону реакции, в результате чего выход изопентана повышается до 97—98%, считая на исходный н-пентан. В этом случае состав непревращенной части сырья совершенно идентичен составу исходного.
Процесс, сопровождаемый возвращением непревращенной (или «условно непревращенной») части сырья в зону реакции, носит название процесс с рециркуляцией. Принцип рециркуляции широко используется в технологии химической переработки нефтяного сырья.
Рассмотрим терминологию и расчетные показатели процесса с рециркуляцией. Непревращенную или «условно непревращенную» часть сырья называют рециркулятом (иногда рисайклом), так как ее возвращают в зону реакции. Отношение количества рециркулята к количеству свежего сырья (принимая суммарную загрузку реактора за 100%) называют коэффициентом рециркуляции, а отношение общей загрузки реактора к количеству свежего сырья— коэффициентом загрузки. Если коэффициент рециркуляции обозначить через К, а коэффициент загрузки через Ки нетрудно установить, что Ki — K+l.
Пример. Допустимый выход изопентана при каталитической изомеризации к-пентана равен 55%. Каковы будут коэффициент рециркуляции и конечный выход изопентана в пересчете на свежее сырье, еслн известно, что выход газа (продукты разложения) составляет 1,5% за однократный пропуск сырья?
Количество непревращенного я-пентана равно: /
100 —(55 + 1,5)= 43,5%
Коэффициент рециркуляции К и коэффициент загрузки Ki равны:
43,5
*=-5бХ = 0'77 *1 = 1.77
Выход изопентана на свежее сырье составляет (55/56,5) • 100=97,3%; остальные 2,7% — газ.
Две последние величины могут быть получены н при умножении выхода за однократный пропуск на коэффициент загрузки:
55-1,77 = 97,3% 1,5-1,77 = 2,7%
Допустимую глубину превращения устанавливают экспериментально. В том случае если состав рециркулята совпадает с составом сырья, коэффициент рециркуляции можно определить сразу после того, как установлена оптимальная глубина превращения сырья.
Если же речь идет об условной глубине превращения, то по сравнению со свежим сырьем термическая или термокаталитическая стабильность рециркулята, как правило, бывает выше. Поэтому если допустимая глубина превращения свежего сырья была хъ то при том же режиме процесса глубина превращения смеси свежего сырья с рециркулятом будет хг, причем X2<Xi. Поскольку глубина превращения снизится, количество рециркулята возрастет. После повторного смешения новой порции рециркулята со свежим сырьем глубина превращения еще снизится — как за счет увеличения доли рециркулята, так и за счет его дальнейшей ароматизации. Глубина превращения х3 меньше Хг, но (хг—*з) меньше (*i—Xz), т. е. с каждым повторным циклом превращения глубина превращения и коэффициент рециркуляции будут все более приближаться к некоторым постоянным величинам, достигаемым при установившемся режиме непрерывного процесса.
При экспериментальном определении коэффициента рециркуляции применительно к сырью сложного углеводородного состава Для практических целей можно ограничиться двумя-тремя последовательными опытами. Например, применительно к термическому крекингу дистиллятного сырья коэффициент рециркуляции устанавливается практически постоянным после двух-трехкратного крекинга252.
Пример. При термическом крекинге фракции 200—350 °С (относительная плотность 0,850) было получено 6% (масс.) газа, 27% (масс.) бензина до 200 °С, 50% (масс.) промежуточной фракции 200—350 °С (относительная плотность 0,890) и 17% (масс.) крекинг-остатка выше 350 °С. Смесь 50% свежего сырья и 50% рециркулята (фракция 200—350 °С от первого крекинга) подвергли повторному крекингу при том же режиме; в результате было получено 5% (масс.) газа, 25% (масс.) бензина, 55% (масс.) фракции 200—350°С и 15% (масс.) остатки; после очередного смешения и крекинга смеси выходы продуктов были следующие: 4,5% (масс.) газа, 24,5% (масс.) бензина, 57% (масс.) фракции 200—350 °С и 14% (масс.) крекинг-остатка.
Считая, что материальный баланс практически стабилизировался, пересчитываем выходы продуктов на свежее сырье (с учетом последнего баланса):
коэффициент загрузки: 100/(100 — 57) = 2,33
24.5-2,33= 57%
4.5-2,33= 10,4% 14-2,33 = 32,6%
выход бензина на свежее сырье: выход газа:
выход крекннг-остатка:
Применение рециркуляции позволяет значительно увеличить выход целевого продукта, но уменьшает пропускную способность
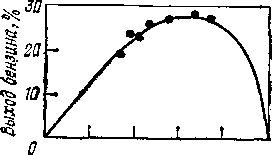
Рис. 1. Зависимость выхода бензина (к.к. 200 °С) от глубины превращения фракции 300—480 °С грозненской пара-финистой нефти при термическом крекинге прн 510 °С и 0,1 МПа.
Данные Г. М. Панченкова и В. Я. Баранова.
0,2 О,if- о,в 0,8 1,9
Глубина. предращещвг%
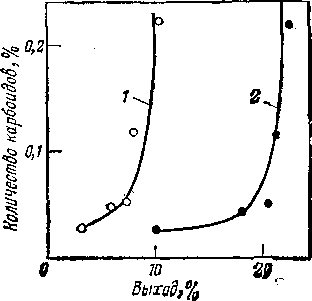
Рис. 2. Зависимость образования карбоидов при термическом крекинге фракции 320—450 °С сернистой нефти от выхода продуктов крекинга:
1 — газ; 2 — бензин.
реактора по свежему сырью. Поэтому допустимую глубину превращения сырья за однократный пропуск нужно выбирать максимально возможной для данного сырья и для данного режима. Допустимая глубина превращения ограничена образованием побочных продуктов — газа или коксовых отложений (продукты уплотнения).
При термическом крекинге фракции 300—480°С грозненской парафинистой нефти снижение выхода бензина (рис. 1) при достижении некоторой глубины превращения объясняется тем, что скорость разложения бензина (до газа) начинает превышать скорость его образования. Из кривой выхода бензина при термическом крекинге фракции 320—450 °С сернистой нефти (рис. 2) видно, что выход бензина за однократный пропуск сырья ограничен образованием карбоидов (продукты глубокого уплотнения): перегиб кривой 2 свидетельствует о том, что в данном случае нецелесообразно увеличивать выход бензина свыше 20%, так как дальнейшее увеличение этого выхода незначительно и сопровождается быстрым возрастанием выхода карбоидов.
При пиролизе газообразных углеводородов после достижения определенной глубины процесса выход целевого продукта (оле-финсодержащий газ) начинает падать, так как реакционноспособная часть газа переходит в жидкие продукты уплотнения. Одновременно возрастает коксоотложение в реакционном змеевике печи, поэтому целесообразно, получив близкий к максимальному выход этилена, после установки газоразделения выделить непре-вращенное сырье и вернуть его в процесс.
ОСОБЕННОСТИ АППАРАТУРНОГО ОФОРМЛЕНИЯ ТИПОВЫХ ТЕРМИЧЕСКИХ И ТЕРМОКАТАЛИТИЧЕСКИХ ПРОЦЕССОВ
Общие принципы устройства реакторов
Учитывая высокие температуры, характерные для термических и значительной части термокаталитических процессов, необходимо вести предварительный подогрев сырья в трубчатых печах.
Для некоторых термических процессов трубчатая печь является одновременно и реактором: в начальной части змеевика осуществляют нагревание сырья до температуры реакции, а остальной участок труб служит для компенсации затрат тепла на крекинг. Если температура термического процесса умеренная (480—520°С) и время реакции измеряется минутами (термический крекинг под давлением) и даже часами (замедленное коксование), то тепло, аккумулированное частично превращенным сырьем в нагревательно-реакционной печи, используется затем для дальнейшего углубления процесса в выносной реакционной камере без внешнего обогрева (рис. 3). Затраты тепла на реакцию в подобных камерах сопровождаются снижением температуры реакционной смеси, т. е. камера работает в режиме, близком к адиабатическому.
Специфика конструкции нагревательно-реакционных печей и выносных необо^реваемых камер подробно разобрана в гл. IV применительно к конкретным термическим процессам.
Крекинг в трубчатых нагревательно-реакционных печах протекает при передаче тепла через стенку труб от зеркала горения и от дымовых газов. Однако для тяжелого смолистого сырья (гудрон, крекинг-остаток) возможности его крекинга и даже подогрева до высокой температуры ограничены, так как в результате реакций уплотнения внутренняя поверхность труб покрывается слоем кокса.
Для термокаталитических и отчасти для термических процессов широко используют принцип передачи тепла крекируемому сырью посредством прямого контакта сырья с горячим твердым теплоносителем. Для термических процессов применяют также газообразные теплоносители (водяной пар, водород, углеводородные газы). Твердым теплоносителем для термокаталитических процессов является катализатор, для термических — инертный материал (кокс, песок). Частицы твердого теплоносителя имеют разные размеры — от крупных гранул округлой или цилиндрической формы до мелкого порошка размером 10—100 мкм.
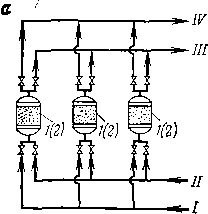
Виды контакта сырья с частицами теплоносителя могут быть различными (рис. 4). На рис. 4,а теплоноситель неподвижен. Его предварительно разогревают, сжигая в отдельном аппарате топливо и подавая в реактор дымовые газы; можно также сжигать в токе воздуха смолисто-коксовые отложения, образующиеся на поверхности теплоносителя в результате процесса, протекающего в реакторе.
Для некоторых каталитических процессов (каталитический ри-форминг) необходимое тепло вносится парами сырья, температура которых по мере протекания процесса в слое неподвижного катализатора снижается, и пары продуктов имеют более низкую температуру, чем вводимое сырье. Этот случай относится к процессам, сопровождающимся эндотермическим эффектом. Если процесс протекает с выделением тепла (гидрокрекинг, гидроочистка), можно обеспечить изотермический режим реактора, снимая избыток тепла холодным водородом. Процессы подобного типа относятся к непрерывным, а катализатор уже не является теплоносителем.
Продукты
' Г
Продуты
у 1 ‘ нагретое\ сырье
Рис. 3. Реакционные камеры без внешнего обогрева: а — с низким уровнем жидкости; б — с высоким уровнем.
Если же проводят периодический разогрев теплоносителя, как указано выше, процесс приобретает полупериодический характер и относится к так называемым сменно-циклическим. Реактор используется по непосредственному назначению только в течение некоторой доли цикла; остальное время затрачивается на подогрев теплоносителя и вспомогательные операции.
Коэффициент использования реакционного объема (по времени) составляет при этом 30—40%.
Если теплоноситель служит и катализатором, то с увеличением размера гранул уменьшается степень использования внутренней поверхности катализатора. Так, для каталитического крекинга при 500°С и диаметре частиц катализатора 3 мм степень использования внутренней поверхности катализатора равна 78%; повышение этой величины до 90% и более потребовало бы уменьшения диаметра частиц до 1,9 мм. Однако, применяя стационарный слой, нельзя брать очень маленькие гранулы, так как при этом резко возрастает сопротивление слоя (рис. 5). Если процесс протекает со значительным тепловым эффектом, соблюдение технологического режима затрудняется недостаточно интенсивной теплопередачей от частиц стационарного слоя к сырью, а также плохой теплопроводностью всей массы теплоносителя. Еще один недостаток описываемой системы — необходимость использования легко-испаряющегося сырья, так как наличие жидкой фазы приведет к неравномерному распределению сырья, к агломерации частиц теплоносителя в результате их слипания и закоксовывания.
Примерами реакционных устройств со стационарным слоем твердого каталитически активного материала являются упомянутые выше реакторы каталитического риформинга, изомеризации, гидроочистки и гидрокрекинга. Применение стационарного инертного материала в качестве теплоносителя весьма мало распространено; в качестве примера можно назвать лишь устаревший ныне
IT
1 111
1У
IV-
¦w
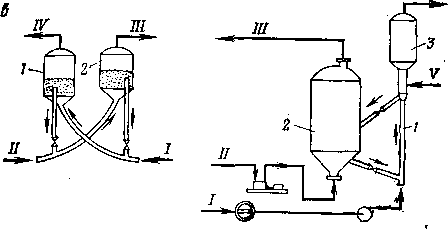
Рис. 4. Реакционные устройства контактного типа:
а — с неподвижным слоем теплоносителя; б — с движущимся слоем крупнограиулироваиного теплоносителя; в — с псевдоожижениым слоем теплоносителя; г *— лифтного типа; / — реактор; 2 — регенератор; 3 — сепаратор; / — сырье; // — воздух; ///— продукты сгорания; IV — продукты реакции; V — водяной пар.
процесс пиролиза легкого нефтяного сырья в газогенераторах и в регенеративных печах.
Реакционное устройство с движущимся твердым теплоносителем представлено на рис. 4,6. В таком реакторном блоке применяют движущийся сверху вниз под действием силы тяжести сплошной поток твердого теплоносителя. Неразрывность потока создается гидравлическим сопротивлением в нижней части аппарата, переходящей в стояк-трубопровод, который выводит теплоноситель в систему пневмотранспорта. Гранулы теплоносителя должны быть крупными (не менее 2 мм) и иметь округлую форму; последнее облегчает их перемещение и сокращает потери от истирания. Сырье можно подавать прямотоком или противотоком к
теплоносителю. Теплоноситель, охладившийся в результате контакта с сырьем, транспортируют в нагреватель (регенератор). Там температура теплоносителя поднимается до первоначальной за счет сгорания кокса, отложившегося на его частицах, или за счет сжигания другого топлива. Теплоноситель нагревается в противотоке воздуха или дымовых газов, поступающих из нижней части нагревателя. Нагретый теплоноситель через второе транспортное устройство возвращают в реактор. Реактор и нагреватель можно располагать по одной оси; при этом устраняется необходимость в одной из линий пневмоподъемника.
Применение движущегося теплоносителя по сравнению со стационарным весьма целесообразно. Функции реактора и нагревателя распределяются между двумя аппаратами, что позволяет спроектировать и эксплуатировать каждый из них наиболее эффективно. Крупнограну-лированный материал движется по основной высоте аппарата равномерно по всему сечению, и только ближе к линии вывода скорость потока частиц в центральной части аппарата увеличивается, периферийные частицы несколько отстают.
0 2 4 6 8
^ Скорость газа, м/с
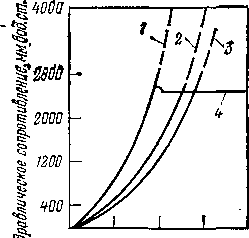
Рис. 5. Зависимость гидравлического сопротивления слоя от скорости газа:
I, 2, 3 — неподвижный слой частиц диаметром 2, 4 и 3—5 мм; 4 — псевдоожиженный слой.
Одинаковая длительность пребывания отдельных частиц теплоносителя в зоне реакции (или нагревания) удобна для контроля технологического процесса. Так, одинаковая степень закоксованности всей массы катализатора при каталитическом крекинге упрощает его регенерацию.
Объемный коэффициент теплопередачи между движущимся слоем теплоносителя и сырьем чрезвычайно велик и достигает 628—2930 тыс. кДж/(м3-К). Температуры теплоносителя и сырья или теплоносителя и воздуха выравниваются чрезвычайно быстро; в условиях промышленных установок этот процесс обычно завершается на протяжении всего нескольких сантиметров по высоте аппарата. Принцип движущегося слоя крупногранулированного теплоносителя используют в процессах каталитического крекинга, пиролиза и некоторых других.
В реакторных устройствах, работающих по принципу «кипящего», или псевдоожиженного слоя (рис. 4, в, стр. 30), твердый теплоноситель находится в виде более или менее тонкого порошка; для каталитического крекинга используют катализатор с частицами от 10 до 120 мкм. Под действием потока газа или паров, Упорядоченного .распределительным устройством (например, решеткой), мелкие частицы теплоносителя приходят в движение, образуя интенсивно перемешиваемый слой, в котором и протекает процесс. Псевдоожиженный слой твердых частиц напоминает жидкость не только по внешнему виду, но и по способности легко перемещаться из одного аппарата в другой по трубопроводам: вниз (под действием силы тяжести) и вверх (с потоком газа или паров).
Взвешенный слой, подобно жидкости, обладает определенным гидростатическим напором. Если высота псевдоожиженного слоя равна h (в м), а плотность слоя равна р (в кг/м3), то давление, оказываемое слоем на решетку, составит:
Лр
юоо (в м В°Д- сг-)
При этом сопротивление псевдоожиженного слоя зависит только от высоты слоя и кажущейся плотности частиц и не зависит от скорости газа и диаметра частиц. Закономерности изменения гидравлического сопротивления неподвижного и псевдоожиженного слоев показаны на рис. 5 (стр. 31).
(Т~* Воздух
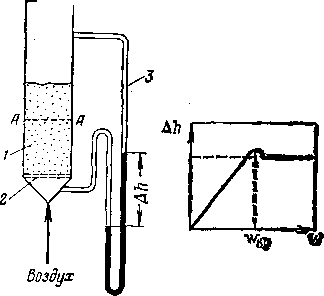
Рис. 6. Схема образования псевдо-ожиженного слоя:
Переход слоя сыпучего материала из неподвижного в псевдо-ожиженное состояние представляет собой качественное изменение. Если сыпучий материал поместить в стеклянный сосуд с пористым днищем (рис. 6) и подавать снизу воздух, постепенно увеличивая его расход и фиксируя показания (ДА) дифференциального манометра, то вначале увеличение расхода воздуха будет сопровождаться почти прямо-. линейным повышением сопротивления слоя. По достижении определенной скорости воздуха, называемой критической скоростью псевдоожижения (taKp), характер слоя изменяется. Частицы слоя под напором воздуха раздвигаются и получают способность перемещаться — слой «закипает»; критической скорости псевдоожижения соответствует максимум перепада давления в слое253. При дальнейшем увеличении расхода воздуха
1 — псевдоожиженный слой; 2 — решетка; 3 — дифференциальный манометр; А—А — начальный уровень слоя (неподвижного).
сопротивление слоя уже не увеличивается, а остается постоянным и равным массе слоя, приходящейся на единицу сечения аппарата.
Псевдоожиженный слой характеризуется высокой интенсивностью перемешивания частиц и значительной теплопередачей от слоя к газу или наоборот. Интенсивность теплопередачи конвекцией зависит от скорости омывания твердых частиц газом, т. е. теоретически она могла бы быть одинаковой для стационарного и псевдоожиженного слоев (при одной и той же относительной скорости потока), но состояние псевдоожижения более благоприятно для контакта частиц с газовым потоком, который распределяется более равномерно. Кроме того, большое значение приобретает перенос тепла за счет теплопроводности псевдоожиженных твердых частиц; для частиц неподвижного слоя, особенно пористых, этот фактор очень мал. В итоге коэффициент теплопередачи в псевдо-ожиженном слое весьма значителен—он составляет от 1047 до 1673 кДж/(м2-ч-К), т. е. 250—400 ккал/(м2-ч-°С).
Вследствие интенсивной массо- и теплопередачи в псевдоожи-женном слое можно обеспечить в реакторе практически изотермический режим, что весьма существенно для большинства процессов и упрощает регулирование режима. Так, для крупных промышленных реакторов каталитического крекинга, имеющих диаметр до 10 м и высоту слоя 5—6 м, температурный градиент по всему объему слоя не превышает 2—3°С.
В реакторе лифтного типа (рис. 4, г, стр. 30) контакт сырья с теплоносителем осуществляется в вертикальной или наклонной трубе. Подобное устройство целесообразно для тех случаев, когда необходимо обеспечить короткое время контакта — до нескольких секунд. Размер частиц теплоносителя при этом обычно невелик — приближается к размеру частиц систем с псевдоожиженным слоем. Реактор лифтного типа можно рассматривать как подъемную линию системы пневмотранспорта- Пары сырья движутся прямотоком с частицами теплоносителя, однако вследствие «скольжения» твердых частиц происходит их некоторое отстаивание. Реактор работает при режиме, отступающем от режима идеального вытеснения, поскольку более свежие порции сырья вступают в контакт с катализатором или инертным материалом, которые прошли тот же путь, что и данная порция паров за более длительное время. Реакторы лифтного типа широко используют в системах каталитического крекинга с мелкодисперсным катализатором и в некоторых модификациях процесса пиролиза.
По гидродинамическому режиму реакторные устройства подразделяются на реакторы идеального вытеснения и реакторы идеального перемешивания. В свою очередь, каждый из этих реакторов может работать периодически и непрерывно. Поскольку в современной нефтеперерабатывающей промышленности реакторы периодического действия практически не используют, ниже рассмотрены только реакторы непрерывного действия.
В реакторах идеального вытеснения время пребывания всех частиц реакционной смеси в зоне реакции одинаково и равно расчетному времени пребывания всей смеси, т. е. сырье, проходя через реактор, непрерывно и постепенно изменяет свой состав от исходного до конечных продуктов реакции. При этом состав реакционной смеси одинаков по всему поперечному сечению аппарата. С некоторой степенью приближения можно отнести к реакторам идеального вытеснения трубчатые печи, реакторы с неподвижным и движущимся слоем крупнограчулированного катализатора (или инертного теплоносителя).
В аппаратах идеального перемешивания поступающее сырье полностью распределяется в продуктах реакции путем механического перемешивания (мешалки непрерывного действия в реакторах сернокислотного алкилирования изобутана олефинами) или в условиях псевдоожиженного слоя мелкогранулированного материала. В последнем случае время пребывания отдельных твердых частиц неодинаково, а пробы паров, взятые из любой точки реакционного объема, имеют одинаковый состав.
Промышленные реакторы отвечают данному разделению лишь с некоторой степенью приближения. Например, в трубчатых реакционных печах для соблюдения режима идеального вытеснения должен существовать так называемый поршневой режим, т. е. должны быть равны линейные скорости всех элементов потока. При существующем обычно турбулентном режиме эпюра распределения скоростей по диаметру трубы отличается от идеальной: скорости по периферии трубы несколько меньше. При прямоточном движении сырья и крупногранулированного материала в реакторе .колонного типа скорость твердых частиц в осевой части аппарата с приближением к его низу возрастает; в результате равномерное движение реакционной смеси и соответственно глубина ее превращения также несколько нарушаются.
Основные параметры работы термокаталитических реакторов
При определении полезной тепловой нагрузки нагревательно-реакционной печи термического крекинга или пиролиза помимо количества тепла, идущего на нагрев и полное или частичное испарение сырья и продуктов превращения, следует учитывать эндотермический эффект реакции крекинга. Таким образом, полезная тепловая нагрузка печи Qnол составит:
Спол = Qe Qp (О
где Qc — тепло, затрачиваемое на нагрев и испарение сырья, кДж/кг; Qp — тепло, затрачиваемое на реакцию крекинга, кДж/кг.
Уравнение (1) должно быть уточнено исходя из того, что состав продуктов крекинга отличается от состава сырья. Таким образом имеем:
Qc = (<7пр — 9с) (2)
где Gc — загрузка печи, кг/ч; ?пр — средняя энтальпия продуктов превращения, кДж/кг; е/с — начальная энтальпия сырья, кДж/кг.
Помимо определения расхода полезного тепла существенным элементом технологического расчета печи является кинетический расчет реакционного змеевика. Если необходимая глубина разложения сырья определяется температурой tp и временем реакции т (в секундах), то объем реакционного змеевика V составит (в м3):
V = vx
где v — средний объем продукта, проходящего через реакционные трубы, м3/с.
Для небольшого участка змеевика, в пределах которого можно предположить линейное изменение температуры (или даже для всего змеевика, если повышение температуры в нем выражается всего несколькими градусами), можно принять, что средняя температура реакции равна:
*н + *к
гр — 2
где t„ и tK — соответственно температуры на входе и на выходе из данного участка труб.
Изменение температуры по длине змеевика (профиль температуры) зависит от конструкции печи, принятой в соответствии с назначением данного термического процесса.
О роли выносной реакционной камеры, в которой используется тепло, полученное сырьем в нагревательно-реакционной печи, сказано выше (стр. 28).
Если назначением реакционного аппарата является углубление процесса при минимальном коксообразовании, как, например, на установках термического крекинга под давлением, то ввод сырья частично превращенного в печи, расположен в верхней части камеры, а вывод продуктов — в нижней (см. рис. 3, а, стр. 29). Это обеспечивает небольшой объем и, следовательно, малое время пребывания наиболее легко коксующейся жидкой фазы продукта, а основной объем камеры заполнен парами. При этом в камере образуется 20—30% общего количества бензина.
Если термическому превращению подвергается тяжелое, легко коксующееся сырье, то в нагревательно-реакционной печи допускается небольшая глубина превращения (чтобы не закоксовывать трубы), а основная роль отводится необогреваемой выносной реакционной камере (процесс замедленного коксования). При этом камера работает с высоким уровнем жидкости, так как частично превращенное сырье входит в нижнюю часть камеры, а продукты реакции уходят сверху (см. рис. 3,6). В результате длительного пребывания при высокой температуре жидкость постепенно превращается в кокс, а газообразные продукты разложения покидают камеру.
В обоих случаях суммарный тепловой эффект реакции отрицателен, поэтому температура на выходе из необогреваемой выносной камеры всегда ниже, чем на входе254. При этом перепад температур тем больше, чем выше относительная доля реакций разложения.
Характеризуя теплоноситель, необходимо указывать его структуру: частицы теплоносителя мо~ут быть пористыми или непористыми. Чем больше пористость, тем при данной плотности вещества меньше насыпная плотность частиц (т. е. масса единицы объема). С пористостью частиц связано также понятие их кажущейся плотности (плотность, при определении которой в объем частицы включен объем, занимаемый порами). Для непористого вещества кажущаяся плотность совпадает с истинной (т. е. с плотностью самого вещества); для пористых веществ эти показатели могут сильно различаться. Так, для типичных алюмосиликатных катализаторов крекинга кажущаяся плотность составляет 1200— 1300 кг/м3, истинная плотность равна 2200—2400 кг/м3, а насыпная плотность не превышает 800 кг/м3.
Насыпную плотность сыпучих материалов определяют взвешиванием известного объема свободно насыпанных частиц (насыпная плотность без уплотнения) или взвешиванием того же объема, но после некоторого уплотнения частиц— обычно путем легкого постукивания донышка мерного цнлнндра о поверхность, на которой он установлен (насыпная плотность с уплотнением). Кажущуюся и истинную плотность гранул можно получить, определив пористость одним из существующих методов255.
При определении истинной скорости газа в слое необходимо знать порозность слоя, т. е. относительный объем, занимаемый пустотами. Порозность свободно насыпанного слоя е равна:
где у» — кажущаяся плотность частиц; уИ — насыпная плотность частиц. ¦
Таким образом, если скорость паров или газов в свободном сечении аппарата равна Wo, истинная скорость их в слое теплоносителя составит:
Щ
W = ~T
Для реакторов с неподвижным или с движущимся слоем круп-ногранулированного теплоносителя (или катализатора) необходимую загрузку твердых частиц определяют как известный для данного аппарата объем, умноженный на насыпную плотность твердого материала-
Для расчета основных показателей реакторов с подвижным слоем катализатора необходимо знать как основные гидродинамические показатели сыпучего слоя материала, так и специфику протекания реакций в этих системах.
К основным гидродинамическим показателям слоя относятся критическая скорость псевдоожижения (или скорость начала псевдоожижения), определяющая минимальный расход газа на псевдоожижение, и скорость витания частиц, определяющая минимальный расход газа на пневмотранспорт частиц.
Для определения критической скорости псевдоожижения предложено большое число формул. Во все эти формулы в той или иной модификации входят диаметр твердых частиц или их кажущаяся плотность, плотность и вязкость той газовой среды, где происходит псевдоожижение. Хорошую сходимость с экспериментальными данными дает преобразованное уравнение О. М. Тодеса:
Аг
ReK — 1 _ е
Ё
где ReK — параметр Рейнольдса для критической скорости псевдоожижения, равный Re„ = wi/v (и — скорость газа в свободном сечении аппарата, м/с; d-^ средний диаметр твердых частиц, м; v — кинематическая вязкость газа, м2/с); Аг — критерий Архимеда, равный
gd3 (V — Vo) v2Yk '
У — кажущаяся плотность твердых частиц, кг/м3; е — порозность слоя; ук — плотность газового потока, кг/м3.
Порозность неподвижного слоя обычно равна 0,4. Порозность слоя в начале псевдоожижения, когда слой слегка расширен, может быть принята равной 0,5. Некоторую условность в существующие формулы вносит величина d, если твердый материал представляет собой полидисперсную смесь, т. е. смесь частиц широкого гранулометрического состава. Средний диаметр определяют по формулам
d = 2 Xidi (5)
где Xi —массовая доля частиц, имеющих диаметр dt.
И. М. Разумов рекомендует при определении потери напора в слое сыпучего материала использовать формулу (4), которая дает несколько более низкие значения d, так как потеря напора с уменьшением диаметра частиц возрастает, и формула (4) обеспечивает некоторый запас.
0,2в,кг[(мг-ч)
зооо-^ат d,m
^ 1,25
р,нПа-с(сПз)
ОЩ-п
14
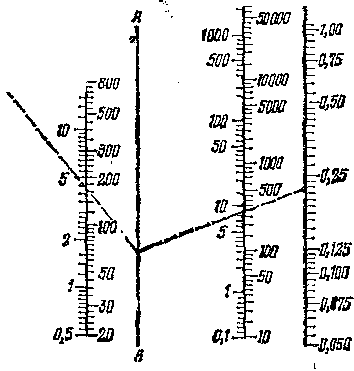
Рис. 7. НоМограмма для приближенного определения критической скорости псев-доожижения (данные М. Лева).
А — А — вспомогательная шкала.
OjOZSi
та,
т-
т:
Для приближенных расчетов критической скорости псевдоожижения можно рекомендовать номограмму, изображенную на рис. 7.
Пример пользования номограммой показан пунктирными линиями. На крайней слева шкале отложена вязкость ц ожижающего газа при рабочих температуре и давлении; на второй левой шкале откладывают величину 0,004 (y—Yo)Yo, где y — кажущаяся плотность частиц (кг/м3), а \о — плотность газового потока (кг/м3). Крайняя справа шкала указывает средний диаметр частиц d (мм); для широкой фракции его определяют как средневзвешенный. Скорость псевдоожижения G (вторая шкала справа) определяют как массовую скорость потока, отнесенную к свободному сечению аппарата и выраженную в кг/(м2-ч).
Для определения линейной скорости полученную величину следует разделить на 3600 Yo-
Пример. Определить критическую скорость псевдоожижения для фракции порошкообразного кокса со средним размером частиц 0,2 мм и плотностью 1700 кг/м3 в среде дымовых газов при 550 °С и «0,14 МПа (избыток кислорода и оксид углерода в продуктах сгорания практически отсутствуют).
Продукты полного сгорания нефтяного кокса, представляющего собой на 97% углерод, будут состоять в основном из азота (азот из воздуха) н диоксида углерода. Простейшим подсчетом можно установить, что в продуктах сгорания содержится 71,0% (об.) азота и 27,0% (об.) диоксида углерода; остальные 2,0% (об-) ~ водяной пар:
Средняя плотность газа равна
28,0-71 +44-0,27+ 18-0,02 --22~4- = ’ ^
а в работах условиях она составит:
1,43-1,4-273
-g2g-= 0,66 кг/м3
Вязкость газа определяют по справочникам256. При 550 °С динамическая вязкость азота равна 3,533-10-5 кг/(м-с), а для диоксида углерода 3,468-10-5 кг/(м-с). Вследствие близости этих значений вязкость дымовых газов условно принимаем равной 3,5-10-5 Кг/(м-с), т. е. 0,035 сПз.
При определении произведения (у—Yo)Yo вследствие большой разницы в плотности твердых частиц и газа величиной уо в первом множителе можно пренебречь. Отсюда имеем:
VY0= 1700-0,66= (1122 кг/м3)2
Для использования номограммы, изображенной на рис. 7, умножаем 1122 на 0,004 и получаем 4,5. Соединяя прямой отложенные на левых шкалах номограммы значения р.=0,035 мПа-с и уу0-0,04 = 4,5, а также точку пересечения прямой на вспомогательной шкале Л—Л с точкой, соответствующей диаметру частиц 0,2 мм на правой шкале, получаем 0,2 (3=9, т. е. (3=45 кг/(м2-ч). Для перевода критической массовой скорости начала псевдоожижения в линейную делим полученное значение G на плотность газа и иа 3600:
45
Ш*Р = 0,66-3600 ^ 0,019 м/с
Если исходные даииые выходят за пределы шкал номограммы, можно использовать уравнение, по которому она была построена:
Г/ч? — л? 10,94
G = 0,00923d1 >82 —-х-?~--(6)
г ’
Здесь все обозначения прежние, но d выражен в м, а ц — в кг/(м-с); при этом G получается в кг/(м2-с).
Обычно практическая скорость псевдоожижения намного выше критической; в промышленных аппаратах осуществляют очень интенсивное псевдоожижение при скоростях, превышающих критическую в несколько раз, с целью улучшения теплопередачи и условий контакта, а также для сокращения диаметра аппарата- Отношение практической скорости псевдоожижения газа к критической называется приведенной скоростью псевдоожижения.
Для расчета скорости витания частиц из соображений, изложенных на стр. 38, И. М. Разумов рекомендует формулу (5), дающую несколько более высокие значения искомой скорости.
Для характеристики глубины контактного процесса вводят понятия объемной или массовой скорости подачи сырья, т. е. скорости, с которой сырье поступает на единицу объема или массы теплоносителя. Скорость подачи сырья в реактор измеряют в м3/(м3-ч) или в кг/(кг-ч), т. е. в обоих случаях после упрощения размерность относительной скорости подачи получается ч-!. По-| скольку плотность сырья и насыпная плотность катализатора не] совпадают, численные значения объемной и массовой скорости не равны, и во избежание ошибки следует писать их полную размерность или делать специальную оговорку. Применительно к систе- j мам с псевдоожиженным слоем твердого материала следует указывать скорость на единицу массы частиц, так как объемы их в сво-' бодно насыпанном и псевдоожиженном слоях неодинаковы. ™
Для систем с подвижными твердыми частицами применяют еще понятие кратности циркуляции, означающее массовое отношение циркулирующего теплоносителя (или катализатора) к сырью и выражаемое в кг/кг (безразмерная величина). Легко видеть, что длительность пребывания теплоносителя (или катализатора) в зоне реакции обратно пропорциональна кратности его циркуляции. Если массовая скорость подачи сырья равна ^(ч-1), а кратность циркуляции теплоносителя п (кг/кг), то длительность пребывания теплоносителя в реакционной зоне составит (в ч):
1
При понижении массовой (или объемной) скорости подачи сырья \ процесс углубляется. Если теплоноситель является одновременно ) и катализатором, при увеличении кратности циркуляции длитель- ! ность пребывания катализатора в реакционной зоне сокращается : и тем самым повышается его средняя каталитическая активность. ; Однако высокая кратность циркуляции неэкономична, так как в ] этом случае увеличиваются энергетические затраты на пневмо- 1 транспортирование, ухудшается процесс отпаривания или повы- : шается соответствующий расход пара. •
При расчете реакционных устройств, работающих по принципу | -псевдоожиженного слоя, необходимо знать степень расширения слоя. По аналогии с уравнением (3) для свободно насыпанного слоя, порозностью слоя называют отношение разности кажущейся плотности частиц и плотности псевдоожиженного слоя к кажущейся плотности:
где- Y*—кажущаяся плотность твердых частиц (масса единицы объема), кг/м3.; Yea — плотность псевдоожиженного слоя, кг/м*.
![]()
С повышением температуры критическая скорость псевдоожи-жения, а следовательно, и практические значения приведенных скоростей псевдоожижения уменьшаются. Это можно объяснить преобладающим влиянием на критическую скорость псевдоожижения вязкости газовой среды: вязкость с повышением температуры возрастает в большей степени, чем уменьшается плотность данного газа.
Для мелкоизмельченного материала можно приближенно принять;
Yf2v3
т. е. при повышении температуры воздуха до 400 °С критическая скорость псев-доожнження снизится примерно в 1,4 раза.
Например, при псевдоожижении слоя порошкообразного кокса со средним размером частнц 0,3 мм прн комнатной температуре критическая скорость псев-доожиження равнялась 1,5 см/с; в условиях горения прн «800 °С коксовый порошок того же гранулометрического состава переходил в состояние псевдоожижения при скорости газа в свободном сечения аппарата всего 0,68 см/с.
С повышением давления увеличивается вязкость газа и тем снижается критическая скорость псевдоожижения. Исследование режима псевдоожижения показало, что влияние давления особенно заметно до 5—6 МПа и возрастает для более крупных частиц, критическая скорость псевдоожижения которых соответствует турбулентному режиму потока. Например, для частиц катализатора диаметром 0,67 мм критическая скорость псевдоожижения при атмосферном давлении была равна 0,47 м/с, а при 1 МПа — гораздо меньше (0,27 м/с); в то же время критическая скорость псевдоожижения частиц размером 0,2 мм практически не зависела от давления*.
Каждому значению порозности слоя соответствует его определенная плотность. Однако даже при больших приведенных скоро-' стях объем слоя увеличивается незначительно. При умеренных скоростях газа граница слоя вполне отчетлива, и слой может быть охарактеризован как плотный. С увеличением скорости газа унос частиц увеличивается, граница слоя стирается и плотность его снижается; при некоторой скорости газа его поток преодолевает силу тяжести частиц, и процесс псевдоожижения переходит в процесс пневмотранспорта твердых частиц. Для осуществления пневмотранспорта необходимо, чтобы скорость газа была больше скорости витания частиц, т. е. больше той скорости, при которой частица находится в равновесии (парйт или витает) в потоке газа, так как ее вес уравновешивается подпором газа.
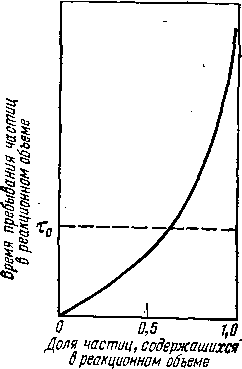
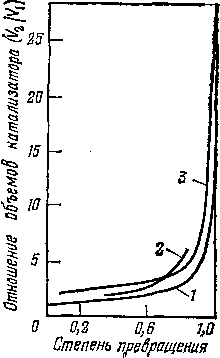
Рис. 8. Зависимость соотношения объемов катализатора в псевдоожиженном слое (Va) и в неподвижном слое (У]) от степени превращения сырья:
1 — реакция первого порядка; 2 — реакция второго порядка; 3 — реакция первого порядка с торможением продуктами.
Рис. 9. Распределение твердых частиц по времени их пребывания в аппарате с псевдоожиженным слоем.
Существенную роль в механизме крекинга в псевдоожиженном слое теплоносителя (или катализатора) играет диффузия газа к наружной поверхности частиц теплоносителя и к внутренней поверхности пор. Исследования псевдоожиженного слоя показали, что в нем происходит перемешивание и твердой и газовой фаз. При этом перемешивание газовой фазы осуществляется в продольном направлении и почти отсутствует в радиальном. В результате состав реагирующей смеси практически одинаков по всей высоте слоя и на выходе из него- Отсюда следует, что для достижения заданной глубины превращения сырья в псевдоожиженном слое объем катализатора должен быть в несколько раз больше, чем в стационарном. Из рис. 8 видно, что объемы стационарного и псевдоожиженного слоев для реакций первого и второго порядка близки при небольшой степени превращения и весьма различаются при углублении процесса. В случае торможения процесса образующимися продуктами разница становится заметной даже при малой глубине превращения.
Для псевдоожиженного слоя характерна неравномерность пребывания частиц в реакционной и нагревательной зонах (рис. 9). Кривая распределения образует с осями графика площадь, равновеликую площади прямоугольника, характеризующего среднее время пребывания теплоносителя в реакторе.
Если процесс каталитический, степень срабатываемости (а в регенераторе — степень восстанавливаемости) разных частиц катализатора неодинакова. Явление перемешивания твердой и газовой фаз может вызвать нежелательные побочные реакции и проскок газов через слой.
Для устранения недостатков псевдоожиженного слоя можно секционировать его на несколько слоев меньшей высоты, а также уменьшать диаметр аппарата и укрупнять частицы. Естественно, что вследствие небольшого размера частиц в системах с псевдо-ожиженным слоем скорость газа, необходимая для их транспортирования, намного меньше, чем для крупногранулированного теплоносителя. Поэтому промышленное оформление транспорта порошкообразных теплоносителей менее громоздко и позволяет иметь установки большей мощности. Подобно реакторному блоку с крупногранулированным теплоносителем, реактор -и нагреватель установок с псевдоожиженным слоем можно располагать параллельно или по одной оси; в последнем случае устраняется одна из ветвей пневмоподъемника.
Реакторы лифтного типа, упоминавшиеся выше, лишены недостатков реакторов с псевдоожиженным слоем — катализатор срабатывается (если речь идет о каталитическом процессе) более равномерно, хотя и наблюдается некоторое перемешивание частиц, летящих по трубопроводу. Основной регулируемый параметр лифт-реактора — кратность циркуляции твердых частиц по отношению к сырью. Зная эту величину, температуру и давление в начале и конце лифт-реактора, можно определить объем газовой фазы и коэффициент взвеси, т. е. число массовых частей твердого материала, приходящихся на одну массовую часть газа- Скорость движения взвеси должна превышать скорость витания самых крупных частиц, а высоту реактора h определяют по простой формуле:
h = wt
где w — линейная скорость движения взвеси, м/с; X — необходимое время контакта твердых частиц с сырьем, заданное режимом процесса, с.
При определении скорости движения взвеси учитывают коэффициент скольжения, представляющий собой отношение средней скорости потока транспортирующего газа к средней скорости твердых частиц.
Процессы с псевдоожиженным слоем теплоносителя широко применяют в нефтеперерабатывающей промышленности. По этому принципу работают реакционные устройства установок каталитического крекинга, коксования, пиролиза, каталитического дегидрирования бутана, гидрокрекинга. Из родственных нефтепереработке процессов следует назвать газификацию угля, синтезы на основе водяного газа. Применение псевдоожиженного слоя особенно облегчает проведение процессов, сопровождающихся значительным тепловым эффектом, который легко компенсируется интенсивным подводом или отводом тепла в слое, а также переработку тех видов сырья, которые трудно нагревать до температуры реакции без их разложения (тяжелые, легко коксующиеся нефтяные остатки).
Исследованию и практическому применению явлений псевдоожижения посвящены многочисленные статьи и монографии (см. рекомендуемую литературу к разд. I).
Погоноразделительная аппаратура
Продукты термических и термокаталитических процессов направляют обычно в систему погоноразделения. В зависимости от вида процесса последовательность и тип погоноразделительных аппаратов различны.
Если требуется выделить из продуктов реакции наиболее тяжелые компоненты, то после реактора (печь или выносная камера)
устанавливают полый цилиндрический аппарат—эвапоратор (испаритель), в котором происходят конденсация и отделение тяжелого остатка. Иногда, для того чтобы обеспечить эту конденсацию, в линию, соединяющую реактор с эвапоратором, подают насосом заданное количество охлаждающего нефтепродукта.
Пары 6 концентрационную часть колонны
Сырье.
Пары из-звапора/пора
Колонны крекинг-установок в основном не отличаются от колонн атмосферной перегонки нефти по типу тарелок, способу подачи орошения, устройству отпар-ных секций для боковых погонов. Однако крекинг-колонны обладают и некоторыми особенностями, вызванными спецификой деструктивных процессов. К этим особенностям принадлежит, в первую очередь, значительный избыток тепла, вносимый в колонну парами из реактора или из эвапоратора. Например, на установках каталитического крекинга в колонну входят пары продуктов, нагретые до 500—520°С, при замедленном коксовании —пары из коксовых камер с температурой «450°С, на установках термического крекинга — пары из эвапоратора,, имеющие температуру «420°С.
Легкая
флегма
Тяжелое а/рье рециркулят
Рйс. 10. Нижняя часть крекинг-колои-яы (с паровым питанием).
На рис- 10 изображена нижняя часть крекинг-колонны, работающая как конденсатор смешения: пары продуктов из эвапоратора встречаются с потоком жидкого сырья. Поскольку температура паров значительно выше, чем температура сырья, происходаг
Жирный 3 г—— гдз
/ L
Жирный | га? а
Вт
1 Орошение
ный бензин
Насыщенный ' аоспрбвнт
^5
Пар
Абсорбент на ГФУ
Z
Орошение
Легкий.
газойль
<_^г\шлана.
Ошстой
?+§
¦ >? 2,
Пар ,8 (E<LD
J L
7
йода
Пары из реактора
Тяжелый газойль,
Пар
Рис. 12. Газосепараторы высокого (1) и низкого (2) давления.
Нестабильный бензин
Жирный I газча прим -I—. компрессора
! Водородсодержащий t ^ I газ
Нестабильный
бензин
Arc
Шлам 5ре а пт of;
Рис. 11. Система утилизации тепла боковых и иижиего погонов ректификационной колонны иа установке каталитического крекинга:
1 — колонна; 2 — конденсаторы и холодильники; 3 — газосепаратор; 4 — насосы; 5 — отпар-ная колонна; 6 — теплообменники; 7 — отстойник катализаторного шлама; 8 — парогенератор. '
нагрев и даже частичное испарение сырья при одновременной конденсации наиболее тяжелых продуктов; в итоге в нижней части колонны находится смесь утяжеленного (или только нагретого) сырья и рециркулята, а сверху уходят пары облегченного фракционного состава.
На установках термического крекинга под давлением применяют комбинированные колонны. Их нижняя часть служит аккумулятором для смеси тяжелого сырья и рециркулята, со сборной тарелки отбирают легкую газойлевую фракцию (питание печи легкого сырья), а сверху уходит смесь паров бензина и газа.
Если в колонну сырье не подают, нерационально снимать избыток тепла только верхним орошением колонны, и практикуется подача промежуточного циркуляционного орошения в нескольких сечениях колонны. Тепло, отнимаемое циркуляционным орошени-ем, используют для подогрева сырья в теплообменниках или для получения водяного пара. Для крекинг-колонн характерно также отсутствие отпарной секции для остатка, так как последний либо направляют на крекинг, либо отводят как побочный продукт, не требующий четкой ректификации.
На рис. 11 представлена промышленная колонна каталитического крекинга. При отводе только одного бокового погона (легкий газойль) в колонне имеются три промежуточных орошения, причем вырабатывается водяной пар трех параметров: 0,3, 0,8 и 1,2 МПа. В нижней части колонны содержатся только каскадные тарелки для осуществления эффективного контакта паров н циркулирующего остатка.
Все процессы деструктивной переработки нефтяного сырья сопровождаются газовыделением, поэтому для многих термических и термокаталитических процессов характерны газосепараторы (рис. 12). Их назначение — отделить жидкий продукт (чаще всего бензин) от газа.
В зависимости от назначения давление в газосепараторах бывает различным. Чем выше давление в газосепараторе, тем легче газ, отделяющийся от жидкой фазы, и тем больше растворено в жидкости более тяжелых газообразных Компонентов. Например, в системах каталитического риформинга, гидроочистки и гидрокрекинга, протекающих в атмосфере циркулирующего водорода, первый по ходу газо-жидкостной смеси газосепаратор служит для отделения водородсодержащего газа, в котором концентрация водорода достигает 90% (об.), а остальные 10% (об-)—в основном метан и этан. Катализат, содержащий растворенный углеводородный газ, перепускают во второй газосепаратор, где давление ниже и где за счет перепада давления из катализата выделяется большая часть растворенного газа. Схема соединения газосепараторов представлена на рис. 12, а.
Давление в первом газосепараторе обычно близко к давлению в реакторе, отличаясь от него только на величину гидравлических потерь в системе теплообмена и конденсации паров. Так, избыточное давление в газосепараторе на установке каталитического крекинга не превышает 0,10—0,15 МПа, поэтому для вывода жирного газа из системы на разделение приходится использовать компрессор (рис. 12,6). В противоположность этому, на установке гидрокрекинга водородсодержащий газ из сепаратора уходит при 15— 17 МПа.
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
Обрядчиков С. Н. Технология нефти. М., Гостоптехиздат, 1952. Ч. II. 408 с. Заородский С. С., Гидродинамика и теплообмен в псевдоожиженном слое. М., Госэиергоиздат, 1963.488 с.
Гельперин Н. И., Айнштейн В. Г., Кваша В. Б. Основы техники псевдоожи-жеиия. М., Химия, 1967. 664 с.
Катализ в кипящем слое/Под ред. Н. И. Мухленова. М., Химия, 1971. 312 с.
Лева М. Псевдоожижение. Пер. с англ./Под ред. Н. И. Гельперина. М.,-Гостоптехиздат, 1961. 400 с.
Разумов М. М. Псевдоожижеиие и пневмотранспорт сыпучих материалов. М.,, Химия, 1972. 240 с.
Брайнес Я. М. Введение в теорию и расчеты химических и нефтехимических реакторов. М., Химия, 1976. 232 с.
РАЗДЕЛ ВТОРОЙ ТЕРМИЧЕСКИЕ ПРОЦЕССЫ
Г .Г1А В А III
ТЕОРЕТИЧЕСКИЕ ОСНОВЫ ТЕРМИЧЕСКОГО КРЕКИНГА НЕФТЯНОГО И ГАЗОВОГО СЫРЬЯ
Основы термодинамики термического крекинга
При современном уровне развития термических процессов сырье для них может быть разнообразным: от низших газообразных углеводородов до тяжелых высокомолекулярных остатков. Поэтому для исследователя и инженера-нефтяника интересно выяснить поведение при высоких температурах самого различного нефтяного и газового сырья, и термический крекинг изучают как на индивидуальных углеводородах, так и на нефтяных фракциях и остатках.
Исследование крекинга углеводородов позволяет получить более строгие кинетические данные и изучить механизм процесса. Эта задача облегчается возможностью четкого отделения продуктов реакции от непрореагировавшего сырья. При крекинге широких нефтяных фракций определить глубину превращения затруднительно, так как сложность химического состава сырья, как сказано выше, не позволяет идентифицировать его непревращенную часть. Однако это обстоятельство не снижает ценности исследований сырья широкого фракционного состава, потому что позволяет определить такой необходимый показатель, как относительную скорость крекинга (т- е. скорость образования бензина, газа, кокса и других продуктов) при различных температурах. Эти показатели могут быть использованы при проектировании и эксплуатации промышленных установок.
Результаты крекинга индивидуальных углеводородов позволяют судить о поведении в этом процессе даже их простейших смесей лишь с некоторым приближением из-за взаимодействия продуктов реакции, а также возможного взаимного торможения этих реакций. В еще большей степени это относится к сложным углеводородным смесям — нефтяным фракциям, при крекинге которых взаимодействие образующихся продуктов и компонентов исходного сырья значительно изменяет состав конечных продуктов превращения, т. е. результат процесса. Поэтому, говоря о крекинге углеводородов какого-либо ряда, обычно имеют в виду начальные
стадии процесса — образование первичных продуктов. При более глубоком крекинге значительную роль приобретают превращения этих первичных и последующих продуктов.
Возможность протекания крекинга определяют исходя из изменения энергии Гиббса (изобарно-изотермический потенциал) дG. Значения стандартных (при 298 К) энергий Гиббса или, что то же, энергий при постоянном давлении, имеются в таблицах термодинамических величин257. Чем ниже AG, тем больше стабильность углеводорода.
Приближенно изменение AG° при любой температуре определяют по уравнению:
AG°r = АН0 —TAS0 (9)
1 298 298 ' '
где ДН 298 — тепловой эффект реакции, вычисляемый по теплотам образования или по теплотам сгорания исходного сырья и продуктов реакции; Т — абсолютная температура реакции; AS§gs — изменение энтропии в результате химической реакции, вычисляемое на основе табличных данных по абсолютным значениям энтропии для компонентов реакции.
Изменение энергии Гиббса связано с константой равновесия реакции:
AG0
1п/Ср= — или AGo=—19,124TlgKp (10)
Константа равновесия равна
k.
— fe
где ki и &2 — константы скорости соответственно прямой и обратной реакций. Если скорость прямой реакции больше скорости обратной, то 6i>&2 и, следовательно, Кр> 1 и lg/Cp>0, т. е. AG0 должно иметь отрицательное значение. Чем меньше абсолютная величина AG0, тем больше термодинамическая вероятность прямой реакции; другими словами, самопроизвольно идущая реакция сопровождается уменьшением изобарно-изотермического потенциала системы.
Для равновесной реакции имеем:
AG° = m1G01+m2(% = ...— — n2Gan (II)
где Gf, G°2, Gf и GJj—изобарно-изотермический потенциал образования соответственно конечных продуктов и исходных веществ; mh m2, щ и Яг—стехиометрические коэффициенты.
•Пример. Определить термодинамическую вероятность алкилирования бензола пропиленом при 25 °С:
С6Н6 4" С,На
ч—— С,Н5
С3Н7
Прн известной энергии Гиббса для бензола, пропилена и изопропилбензола (G&*-146,47 кДж/моль, 0^зн6=77,86 кДж/моль, Оо)Н5с3н7=: ^3,93 кДж/моль) получаем:
183,93- 146,47- 77,86 =-40,40 кДж/моль

Отсюда видно, что алкнлнрованне может протекать при комнатной температуре (25°С), хотя скорость реакции может быть прн этом очень невелика.
Изменение энергии Гиббса при крекинге позволяет установить относительные границы термической стабильности. Для любой реакции Лб°=0 при некоторой температуре t- Так, для н-бутана при его дегидрогенизации при ?«6470С имеем AG° = 0, а при расщеплении этого углеводорода условие AG°=0 соответствует температуре 313 °С. По этим данным легко определить границы возможного протекания реакции и одновременно сделать вывод о преобладающем значении реакций расщепления.
Абсолютные значения энергий Гиббса позволяют также судить о термической стабильности углеводородов: положительные значения свидетельствуют о преимущественной возможности разложения. Так, при 527°С изобарно-изотермический потенциал образования метана равен —2,30 кДж/моль, а для додекана ¦ ,4-669,12 кДж/моль. Известно, что при этой температуре метан практически стабилен, а додекан легко разлагается.
Используя значения энергии Гиббса, можно вывести ее зависимость от температуры при данной реакции, а также найти константы равновесия при любой температуре.
Пусть, например, происходит разложение «-декана на пентан и амилен:
С10Н22 * *' +
С5Н10
Изобарно-нзотермнческне потенциалы образования каждого из этих углеводородов при 298 К соответственно равны (по справочным таблицам): 33,35, 8,37 и 79,33 кДж/моль. Тогда запишем
мышленных установок
(С. Н. Обрядчиков и др.) Калориметрически (Б. К. Америк и др.) Вычислено
(В. Л. Нельсон) То же
То же Обработка промышленных данных (Е. В. Сми-дович)
Вычислено по лабораторным данным (Г. Г. Ва-лявин и соавт.)
AG2gs = —8,37 + 79,33 — 33,35 = +37,61 кДж/моль
т. е. реакция при данной температуре не пойдет. Аналогичные подсчеты для 800 К дают:
ДGg00 — 245,89 + 266,02 — 547,27 = —35,36 кДж/моль
Следовательно, прн 800 К (527°С) реакция термодинамически возможна. Поскольку зависимость изменения энергии Гиббса от температуры имеет линейный характер (AGt=A-\-BT), можно определить по двум значениям ДGt коэффициенты А н В этого уравнения
+37,61 =А + В-298 —35,36 = Л + В-800
откуда А = +80,64 н В=—0,145 а уравнение для данной реакции будет иметь внд:
AGT = 80,64 — 0,1457’
Отсюда Д0т=0 прн 556 К (283 °С), т. е. рассмотренная реакция разложения я-декана термодинамически возможна прн температурах выше 283 °С (при 283 °С скорость разложения ничтожно мала).
Анализ реакций, типичных для деструктивных процессов переработки нефтяного сырья, показывает, что термодинамическая вероятность их протекания возрастает с повышением температуры для реакций разложения и, напротив, с понижением температуры— для реакций синтеза (алкилирование, полимеризация, гидрирование). Это не означает, что все реакции синтеза осуществляют в промышленных условиях при низких температурах, поскольку скорость их протекания может оказаться слишком мала.
В гл. I говорилось о способах определения теплоты реакций, протекающих при химической переработке нефтяного сырья, и о значении этого показателя для проектирования и эксплуатации промышленных установок. Следует иметь в виду, что теплота реакций разложения не зависит от молекулярной массы исходного углеводорода, если отнести ее на 1 моль превращенного сырья. Так, при 500 °С теплота образования этилена из н-бутана и из rt-декана одинакова и составляет ж92 кДж на 1 моль сырья; однако в пересчете на 1 кг сырья эти теплоты составят соответственно 1590 и 650 кДж/кг, что свидетельствует о большей термической стабильности н-бутана.
Теплоту реакции термического крекинга иногда относят к единице массы продуктов превращения (бензин или сумма бензина и
|
Тепловой эффект, кДж/кг (ккал/кг) |
|||
| Процесс | Сырье | на 1 кг сырья | на 1 кг бензина |
| Крекинг под давлением | Газойлевый дистиллят | — |
1250—1460 (298—349) |
| То же | То же и мазут | 1250—1670 (298—399) | |
|
Висбрекинг Пиролиз-дегидрирование Пиролиз Замедленное коксование |
Тяжелые остатки Этан «-Бутаи Тяжелые остатки | 117—234 (28-56) 4460(1070) ж 1250 (и 298) 84—168 (20—40) | — |
|
Периодическое коксование |
Крекинг-остаток высоко^ парафинисто? нефтн | 210(и50,3) |
' |
Способ получения данных
газа). Такой способ выражения условен, так как тепло затрачивается и на образование более тяжелых продуктов разложения, а кроме того, образование продуктов уплотнения сопровождается выделением тепла. Более удобно относить тепло на 1 кг превращенного сырья.
В табл. 1 приведены данные о теплотах реакции основных термических процессов переработки нефтяного сырья.
Основы химизма и механизма термических превращений
Крекинг парафиновых углеводородов. Для крекинга парафинов характерен их распад на более низкомолекулярные углеводороды. Продукты распада состоят из парафиновых и олефиновых углеводородов и водорода.
Термическая стабильность низших, газообразных парафинов очень велика. Так, метан ниже 700—800 °С практически не разлагается. Значительная стабильность метана объясняется тем, что в его молекуле отсутствуют связи С—С, энергия диссоциации которых меньше, чем для связей С—Н. При умеренной глубине разложения метана основными продуктами его крекинга являются этан и водород.
Этан и пропан склонны к реакциям дегидрогенизации с образованием соответствующих олефинов, но для пропана уже при 600 К вероятность разложения на метан и этилен в 1,5 раза больше, чем вероятность дегидрирования до пропилена.
По мере увеличения молекулярной массы исходного углеводорода термическая стабильность его падает, и преобладающими становятся реакции расщепления молекул по связи С—С. Так, к-бутан дегидрируется при крекинге всего на 10%; при этом в качестве основных продуктов образуются смеси метана с пропиленом й этана с этиленом.
Ф. Райс предложил цепной механизм распада парафиновых углеводородов при крекинге. Поскольку энергия связи С—С меньше, чем энергия связи С—Н, то первичный распад молекулы парафинового углеводорода происходит по этой связи и дает радикал, обладающий неспаренным электроном: «СНз, «СгНв, «СзН? и т. д. Продолжительность существования радикалов, более сложных, чем •С3Н7, при температурах крекинга ничтожно мала. Они мгновенно распадаются на более простые, которые могут вступать в реакции с молекулами углеводородов, отнимая у них водород и превращаясь, в свою очередь, в насыщенный углеводород, например:
C2H„ + R- —> .C2h6 + RH
Образовавшийся радикал вступает в реакцию с новыми молекулами углеводородов. Если этот радикал имеет сложное строение, он мгновенно распадается на более простой радикал и непредельный углеводород:
•С6НП—- -ед + СзН,
Радикалы, существующие достаточно продолжительное время, чтобы при данной температуре вступить во взаимодействие с углеводородом (к ним относятся Н-, *СНз и -СгНб), называются свободными.
Рассмотрим последовательность термического разложения парафиновых углеводородов по Райсу на примере н-бутана.
1. Первичный распад молекулы на радикалы. Поскольку дегидрирование здесь менее вероятно, чем разрыв по связи С—С, можно предположить образование двух радикалов:
СН3-СН2-СН2-СН3 -—* 2-С2Н6
2. Развитие цепи. Образовавшийся радикал взаимодействует с молекулой исходного углеводорода:
сн3—СН2—СН2—СН2 + С2Нв СН3—СН2—сн—сн3 + С2Н6
СН3-СН2-СНа-СН3 4- -СаН5
Райс определяет вероятность места отрыва водорода, присоединяющегося к свободному радикалу, исходя из следующих соображений:
а) число одинаковых групп, содержащих водород (в данном
случае две крайних группы *СН3 и две средних группы «СНг);
б) число атомов водорода в группе (три и два для й-С4Ню);
в) относительная скорость взаимодействия радикалов с атомами водорода при первичном (*СН3) и вторичном («СНг) атомах углерода при данной температуре. Водород при третичном атоме углерода наиболее активен.
Относительная скорость взаимодействия радикалов с атомами водорода по Райсу такова:
“С *сн3 -сн2 .сн
Из этих данных видно, что при 600 °С скорость взаимодействия водорода при группе *СН2 вдвое больше, чем при группе *СН3.
Выше показано, что распад я-бутана может протекать по двум вариантам. Первый такой
СНа— СН2—СН2—СН, +«• -> СН,-СН2—CHa-CHs + RH
•но радикал »С4Н9 нестабилен и распадается по связи, отстоящей, по крайней мере, через одну от группы с неспаренным электроном, так как смежная с этой группой связь наиболее прочна:
СН3-СН2-;-СНа-СН2 + RH - С2Н4 + С2Н, + R.
Второй вариант такой:
CH3-CHa-CH2-CHs + R« -> ОТ,—СН—СН2—ОТ, + RH
СН3—СН—СНа—CHS + RH'-> CjHa + СН4 + R-
В итоге первая реакция дает этилен и этан, а вторая пропилен и метан. Вероятность первой реакции по вышесказанному равна
2258 3 -1=6. Вероятность второй реакции составляет: 2 - 2 - 2 = 8, т. е. из четырнадцати молекул и-С4Ню шесть разложатся на С2Н4 и С2Нб, а восемь — на С3Н6 и СН4.
3. Образующиеся радикалы вновь вступают во взаимодействие с молекулами исходного углеводорода, концентрация радикалов возрастает и возникает значительная вероятность столкновения двух радикалов с образованием парафиновых углеводородов или молекулы водорода:
R-+R'- -*¦ R—R' \
R* + Н • -> RH 1 обрыв цепи
Теория радикалов позволяет довольно точно предсказать состав продуктов распада простейших парафиновых углеводородов при давлении, близком к атмосферному.
Крекинг нафтеновых углеводородов. Термический распад нафтеновых углеводородов происходит не по радикально-цепному, а по молекулярному механизму. Некоторые исследователи* предполагают возможность крекинга циклопропана, циклобутана и их производных через стадию образования бирадикалов.
Наиболее вероятным направлением термического разложения алкилнафтенового углеводорода является разрыв алкильной цепи, так как связи С—С в цикле значительно прочнее связей в алкильной группе; при повышении температуры может происходить полное деалкилирование углеводорода с последующим насыщением водородом свободной связи у атома углерода, входящего в цикл.
При более жестком режиме крекинга происходит, кроме того, разрыв кольца с образованием олефиновых или диеновых углеводородов. Бициклические нафтены с алкильными цепями сначала превращаются в полностью или частично деалкилированные наф-
Дегидрогенизация моноциклических нафтенов до соответствующих ароматических углеводородов протекает через стадию образования циклических непредельных углеводородов. Эта реакция свойственна жесткому режиму крекинга (главным образом пиролизу). Так, превращение циклогексана в бензол протекает с отрицательным изменением энергии Гиббса при температурах выше 660 °С. При 622 °С, по данным Ф. Е. Фрея, крекинг циклогексана дал 44,1% олефинов (до С4), 9,5% бутадиена, 3,7% циклопентена и амиленов, 4,9% циклогексена и циклогексадиена и только 0,9% бензола (и 1,2% высших углеводородов).
Бициклические нафтены, например декалин, при крекинге также в основном дают продукты разложения (алифатические углеводороды, моноциклические нафтеновые и ароматические углеводороды) и в меньшей степени — продукты дегидрирования (в данном случае тетралин и нафталин).
тены с последующим разрывом одного из колец и дальнейшим де-алкилированием моноциклического нафтена:
Крекинг ароматических углеводородов. Простейшим представителем голоядерных углеводородов является бензол (т. кип. «80°С). Бензольное кольцо чрезвычайно стабильно, однако бензол довольно легко переходит в дифенил, что сопровождается выделением водорода:
2С6Н6
4.
> СвН6—С6Н6
-j- Н2
Помимо дифенила образуются полифенилы, а также смолистые вещества и кокс. В газах крекинга кроме основного продукта разложения— водорода — содержатся парафиновые углеводороды, что можно объяснить разрывом части молекул исходного бензола. Так как образование дифенила из бензола — обратимая реакция, то при соответствующих условиях (при повышении парциального Давления водорода) равновесие сдвигается в сторону образования бензола (наряду с образованием продуктов конденсации).
Аналогично бензолу ведет себя нафталин. При его крекинге жидкие продукты разложения не образуются, а получаются только продукты конденсации (динафтил) и газ, богатый водородом. Такое направление реакции свойственно и трехкольчатьм ароматическим углеводородам — антрацену и фенантрену. Однако установлено, что некоторые еще более сложные ароматические углево-перегоняется без разложения при весьма высоких температурах (намного выше 500 °С).
М. Д. Тиличеев предложил радикальный механизм распада голоядерных ароматических углеводородов, хорошо совпадающий с результатами экспериментальных исследований. Образование ароматического радикала происходит в результате взаимодействия бензола (нафталина и т. д.) с атомом водорода:
дороды термически стабильны. Так, коронен, имеющий общую формулу С24Н12
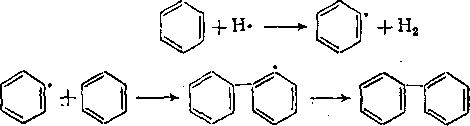
+ Н- ит. д.
Ароматические радикалы вступают в реакции рекомбинации, приводящие ко все большему усложнению структуры образующихся молекул и к обеднению их водородом. Поскольку связи в ароматических кольцах очень прочны, при крекинге алкилароматических углеводородов происходит в первую очередь частичное отщепление алкильной цепи с образованием алкилароматических углеводородов более простого строения. Так, в процессе крекинга цимола (гс-метилизопропилбензол)
СН3-С6Н4-СН(СН3)2
было получено 26% толуола и ксилолов; бензол отсутствовал, т. е. полного деалкилирования не происходило. Для алкилароматических углеводородов характерна конденсация через метильные группы, а не путем соединения бензольных колец. Например, при крекинге гс-ксилола получается п-диксилол:
СН3—C6H.j—СН2—СН2—С6Н4—СН3
Аналогично ведут себя и некоторые полициклические углеводороды с боковыми цепями, например метилнафталин. При высоких температурах крекинга (700—800 °С) может протекать дегидрирование алкильной цепи, например этилбензол частично превращается в стирол.
Крекинг непредельных углеводородов. Нефтяные фракции и остатки от прямой перегонки нефти содержат очень небольшое ко-
личество непредельных углеводородов. Тем не менее поведение последних при термическом крекинге нефтяного сырья представляет практический интерес, так как олефины являются продуктами первичного распада парафинов. Непредельные углеводороды могут также содержаться и в нефтяном сырье вторичного происхождения: в дистиллятах термического и каталитического крекинга, коксования и т. д.
Олефинам свойственны весьма разнообразные термические превращения, направление которых зависит от температуры и давле-. ния. Умеренная температура (примерно до 500°С) и высокое давление способствуют протеканию полимеризации олефинов; напротив, высокая температура и низкое давление вызывают их распад. Так, из простейшего олефинового углеводорода — этилена —при «15 МПа уже при 370°С образовалось 92% жидких полимеров и 8% бутилена. При 625°С и атмосферном давлении выход жидких компонентов снизился до 32,9%; остальное приходилось на газообразные продукты. При повышении температуры до 800—900°С этилен не полимеризуется — идут реакции разложения и частично конденсации в ароматические углеводороды. Такое поведение характерно и для жидких олефинов.
Наряду с полимеризацией и разложением идут циклизация и дегидрогенизация олефинов. Наличие насыщенных углеводородов в продуктах крекинга олефиноз показывает, что при разложении' не только образуются два олефина меньшей молекулярной массы, но протекает реакция перераспределения водорода с образованием системы парафин + диен. Последний, будучи весьма нестабильным, вступает в реакции конденсации с олефинами.
Механизм первичных реакций термического разложения олефинов, как и для парафинов, является радикально-цепным. При этом первичный распад олефина происходит по p-связи, имеющей наименьшую энергию диссоциации. Например, а-амилен будет распадаться на этильный и аллильный радикалы:
ch3-ch2-Uch2-ch=ch2--* -С2Н6 + .С3Н6
Образовавшиеся радикалы встречаются с исходным амиленом:
QHjo+R- ->- RH + СН3—СН2—СН—СН=СН2
Радикал «CsHg нестабилен, и наибольшая вероятность его распада— отрыв метильного радикала:
СН3—СН2-СН—СН=СН2-> С4Н6 + -СН,
развитие цепной реакции
Последующее развитие цепной реакции идет через радикал .СН3:
Для газов пиролиза характерно наличие бутадиена и метана. Бутадиен образуется также и прямым дегидрированием бутилена.
Крекинг сернистых соединений. Значительная доля перерабатываемых в Советском Союзе нефтей относится к сернистым (от 0,51 до 2% S) и высокосернистым (свыше 2% S). Как правило, концентрация сернистых соединений увеличивается с утяжелением нефтяных погонов. Содержание серы в бензиновых фракциях прямой гонки относительно невелико; в газойлях доля сернистых соединений может приближаться к концентрации ее в исходной нефти; в мазутах содержится в 1,5—2 раза больше серы, чем в исходной.
Среди сернистых соединений нефти преобладают тиофены и сульфиды. По данным Р. Д. Оболенцева, при перегонке высокосернистой нефти в светлые нефтепродукты (до 300 °С) попадает около 20% серы, наполовину в виде сульфидов; из остальных 80% серы, концентрирующейся в мазуте, 85—90% составляет «тиофе-новая» сера.
Данных о термической стабильности сернистых соединений нефти и о кинетике их разложения относительно мало. Известно, что некоторые сернистые соединения начинают разлагаться уже при 160—200 °С. Другие, напротив, термически довольно стойки. Так, разложение бензтиофена (изученного в виде 5%-ного раствора в керосине) начинается только при 450 °С.
Разложение большинства сернистых соединений, содержащихся в нефти, сопровождается выделением сероводорода. Сульфиды и меркаптаны, например, разлагаются по реакциям:
RSR' -»- H2S -f- Олефин RSH -*¦ H2S + Олефин
Поэтому газы, получаемые при крекинге сернистого сырья, как правило, содержат сероводород. При разложении сернистых соединений образуются также жидкие органические соединения серы, например меркаптаны, которые переходят в бензиновые фракции крекинга. Возможно также выделение свободной серы.
В промежуточных фракциях и продуктах уплотнения, полученных при термическом крекинге сернистого сырья, содержится значительное количество серы, что указывает на присутствие термически стабильных сернистых соединений. К их числу относятся соединения типа тиофенов, образовавшиеся в результате крекинга или перешедшие из исходного сырья.
Механизм реакций уплотнения. Протекание этих реакций обусловлено присутствием ароматических, непредельных углеводородов и смолисто-асфальтеновых веществ нефти. С углублением крекинга в его продуктах накапливаются, наиболее термически стабильные голоядерные (или с короткими алкильными цепями) по-лициклические ароматические углеводороды, которые вступают в реакции поликонденсации, постепенно обедняясь водородом.
Природные асфальтены имеют молекулярную массу 2000—2500
и представляют собой сочетание полициклических структур — преимущественно ароматических, но дополненных конденсированными нафтеновыми циклами, а также метильными и метиленовыми группами, атомами S, N и О. Строение природных асфальтевов одной из западносибирских нефтей таково:
При термическом воздействии асфальтены разлагаются с образованием газа, жидких масел и кокса. Асфальтены разных нефтей очень неодинаковы по термической стабильности, что объясняется разнообразием их структуры и состава, однако большинство ас-фальтенов довольно стабильно.
Природные асфальтены (из нефти) содержатся только в остаточном прямогонном нефтяном сырье. При крекинге как остаточного, так д дистиллятного сырья образуются асфальтены вторичного происхождения, значительно отличающиеся от природных. Молекулярная масса их ниже, чем у природных, и тем меньше, чем глубже протекал крекинг. Так, для одного и того же сырья (смесь татарских нефтей) асфальтены, содержащиеся в мазуте, имели молекулярную массу ?«2500, а асфальтены, содержащиеся в остатке после неглубокого крекинга гудрона, — всего 1300. Асфальтены, выделенные из смол пиролиза, в зависимости от жесткости процесса имели еще меньшую молекулярную массу (330—380).
Пониженная по сравнению с природными молекулярная масса вторичных асфальтенов объясняется менее сложными составом и структурой их молекул. Элементный состав асфальтенов различного происхождения представлен в табл. 2. Более высокое содержание углерода во вторичных асфальтенах свидетельствует о значительном преобладании в них ароматических структур и о малом содержании боковых алкильных цепочек.
Начало образования продуктов уплотнения зависит от состава исходного сырья и режима крекинга. Сырье, содержащее парафиновые и алкилароматические углеводороды, претерпевает вначале разложение, подготавливающее материал для последующих реакций уплотнения; таким материалом являются голоядерные ароматические и непредельные углеводороды. Образование продуктов уплотнения происходит по радикально-цепному механизму через алкильные и бензильные радикалы. Последовательность и тип образующихся продуктов уплотнения ясны из схемы:
Алкилароматические Чафтены Парафины
углеводорода
одороды I 1
Го-ПРндерные ароматические Непредельные
углеводороды углеводороды
дет. одор
йвлициклические Алкенил-
Ароматические ароматические
USneBpVopgdbt уелеВодорйЗы
......—
>Смолы •< —*— -*
'
+
Асфальтены
ftapdeHU
Нарбоидь.
Каждый последующий продукт уплотнения обладает все более высокими молекулярной массой и степенью ароматичности, а также уменьшающейся растворимостью. Карбоиды не растворимы в горячем бензоле; карбены растворимы в бензоле, но нё растворимы в сероуглероде и хлороформе; асфальтены растворимы во всех этих растворителях, но осаждаются легкими парафинами.
В промышленных условиях при жидкофазном крекинге в качестве конечного продукта образуется твердое углеродистое вещество— кокс. Основная его масса представлена карбоидами, но в процессе образования в коксе могут частично оставаться непре-вращенные продукты менее глубокого уплотнения — карбены, асфальтены и даже наиболее тяжелые углеводороды. Кокс может являться целевым продуктом, и в этом случае стремятся получить его максимальный выход (коксование). В других случаях образо-
Таблица 2. Асфальтеиы природного и вторичного происхождения
| Мол. масса |
Плот | Элементный состав, % | Отно | ||||
|
Происхождение | ность, КГ/мЗ | С | н | S | N + о |
шение с:н | |
| Нефть Западной Сибири* (Мамонтовская) |
2080 | 1178 | 86,52 |
7,59- | 5,38 | 0,51 | 11,4 |
|
Самотлорская нефть | 2670 | 1092 | 88,70 |
8,69 | 2,00 | 0,61 |
10,2 |
| Смесь татарских нефтей** | 2500 |
— | 86,86 | 8,69 | 4,45 | Не определяли | 10,0 |
|
Крекинг-остаток гудрона той же смеси Смола, получаемая при пиролизе керосина*** | 1300 | 88,72 | 6,96 | 4,32 | 12,7 | ||
|
мягкий режим | 380 |
— | 93,37 | 6,37 |
Нет | 0,26 | 14,7 |
| жесткии режим |
330 | 93,83 |
5,42 | » | 0,75 |
17,3 | |
* Данные Л. Е. Свиитицких.
** Данные Н. С. Казанской и Е. В. Смидович.
*** Данные О. Ф. Глаголевой, Л. И. Сосулииой и Е. В. Смидович.
вание кокса нежелательно; например, при пиролизе е целью получения газообразных олёфинов в трубчатом реакторе последний может закоксовываться, и в результате пробег установки сокращается.
Основы кинетики термических процессов
Чтобы установить оптимальные режимы термодеструктивных процессов переработки нефтяного сырья, необходимо располагать данными о кинетике соответствующих реакций, причем важно, чтобы эти данные были получены в условиях, моделирующих промышленный процесс.
Если промышленный процесс оформлен как полностью непрерывный, т. е. с непрерывной подачей сырья и непрерывным выводом продуктов, изучение его в периодически действующем аппарате (например, в лабораторном кубе) может дать только приближенные данные о материальном балансе и качестве получаемых продуктов. Что же касается данных по кинетике процесса, т- е. получения зависимости глубины превращения сырья от температуры и времени, то эти сведения будут еще более условны. Последнее объясняется тем, что при периодическом процессе продукты непрерывно уходят из зоны реакции, а непрореагировавшее сырье вместе с продуктами первичного разложения и уплотнения остается в жидкости (в аппарате). При этом температурный редким и особенно время пребывания сырья в зоне реакции, как правило, не будут совпадать с заводскими и могут быть сопоставлены с ними только условно, если использовать в качестве критерия глубину разложения сырья или выход целевого продукта.
Изучение кинетики превращения сырья в аппарате периодического действия, однако, может в некоторых случаях дать интересные и надежные результаты. Для этого необходимы два условия: 1) проведение крекинга с небольшой глубиной превращения сырья и 2) использование сырья тяжелого фракционного состава. При небольшой глубине превращения можно установить кинетику первичных реакций разложения сырья, не осложненных еще частичным испарением продуктов, вторичными реакциями распада и уплотнения. С другой стороны, использование тяжелого сырья обеспечивает минимальное испарение и сырья и продуктов его первичного распада.
На лабораторной проточной установке сырье проходит через реакционную зону непрерывно, при постепенном возрастании глубины его превращения, т. е. в этом отношении процесс полностью воспроизводит промышленный. Однако и в этом случае имеются условия, несоблюдение которых может исказить получаемые результаты. К числу их относится гидравлический режим потока — как правило, турбулентный в реакционных змеевиках промышленных печей и часто ламинарный на лабораторных установках. Это обусловливает в последнем случае возможность местных перегревов стенки, приводящих к побочным реакциям разложения и уплотнения. Значительную роль играет также пристеночный эффект, определяемый соотношением внутренней поверхности реакционного змеевика и его объема. Влияние этого фактора, естественно, тем больше, чем меньше масштаб установки; оно зависит от материала стенки и может быть устранено использованием, например, кварцевого стекла. Указанные условности кинетических данных, полученных в лаборатории, не умаляют значения подобных исследований.
Кинетика первичных реакций термического крекинга при относительно небольшой его глубине приближенно подчиняется уравнению мономолекулярного превращения
~=k(a~x) (12)
которое после интегрирования дает:
![]()
где k — константа скорости реакции, с-1; а — количество исходного сырья, моль/с; х — глубина превращения сырья, моль/с; т — время, с. 1
По этому уравнению можно обрабатывать результаты термическо! го крекинга нефтяного сырья, полученные в реакторах периодиче* ского и непрерывного действия, если глубина превращения сырья относительно невелика. Константы скорости крекинга для некоторых нефтяных фракций, вычисленные по уравнению мономолеку-лярной реакции, приведены в работах М. Д. Тиличеева, С. Н. 06-рядчикова, В. J1. Нельсона и др.
А. И. Динцес и А. В. Фрост показали, что при углублении процесса константа скорости реакции уменьшается. Это явление они объяснили тормозящим влиянием продуктов разложения на основную реакцию (такое самоторможение происходит вследствие обрыва реакционной цепи образующимися продуктами). Эти исследователи предложили для крекинга другое уравнение
dx k (а — х)
dx ~~, a —(а —х)
имеющее после интегрирования такой вид:
fe = 4~(alnT3T-^) <15)
Здесь р — постоянная, характеризующая степень торможения, а остальные обозначения прежние.
Подобное уравнение позднее было выведено теоретически А. Д. Степуховичем на основании радикально-цепного механизма крекинга.
Для реакций в потоке (на установке непрерывного действия) при увеличении глубины разложения сырья объем образующихся продуктов все более превышает объем паров сырья, что следует учитывать при определении длительности реакции. Г. М. Панчен-ков предложил кинетическое уравнение, которое описывает термический крекинг как реакцию первого порядка, протекающую в потоке (в соответствии с режимом промышленных установок):
6— 1
п0х = —jj—п0 In (1 — х) + k' (16)
где п0 — число молей исходного вещества, поступающего в зону реакции за единицу времени; k’ — относительная константа скорости, равиая:
PV
к'=к‘Щтг P = v1 + v2 + vs + ...—1
Vi — стехиометрические коэффициенты.
Величину k' удобно определято графически, откладывая по оси абсцисс величину —гс01п(1—х), а по оси ординат п0х (рис. 13). Тогда построенная прямая линия отсекает на оси ординат величину k'. Прямые, построенные для разных температур, должны быть параллельны друг другу. Экспериментальная проверка уравнения (16) показала, что полученные прямые параллельны практически До температур 570—580 °С, являющихся максимально возможными при промышленных процессах термического крекинга и коксования.
Уравнение (16) применимо для такого сырья, которое в условиях крекинга находится в газовой фазе, и, следовательно, для промышленных крекинг-установок, предназначенных для легкого
сырья (реакторы пиролиза, глубокого крекинга газойлей, термического риформинга бензино-лигроиновых фракций).
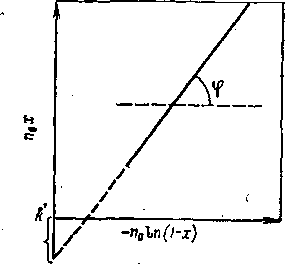
Рис. 13. График зависимости ПоХ от —я01п(1—х) для реакции первого порядка, протекающей в потоке.
Наиболее точное представление о кинетике такого сложного процесса, как термический крекинг, дает его по-стадийное изучение, так как если на первых стадиях преобладают (и являются определяющими) реакции разложения, то в последующем начинает протекать взаимодействие продуктов крекинга, в частности уплотнение, подчиняющееся уравнениям реакций выше первого порядка. В табл. 3 приведены константы скорости, вычисленные для крекинга нефтяных фракций.
Константы скорости, вычисленные для каких-либо двух температур, позволяют рассчитать энергию активации крекинга, пользуясь уравнением Аррениуса:
Таблица 3. Коистаиты скорости крекнига и энергия активации (при атмосферном давлении)
Данные Г. М. Панченкова н В. Я. Баранова
| 10»А, | с-1 | ||||||
| Нефтяная фракция |
при 470 °С | при 490 "С |
ири 510'С | прн 530 “С |
при 540 °С | при 550 *С |
Е, кД ж/мол |
|
300—360 °С (Pt° =0,840)* 300—480 °С (р|о =0,885)** 320—450 °С (pf* =0,896)*** | 0,49 | 1,90 | 1,73 3,20 1.10 | 4,64 | 10,40 4,36 |
10,28 | 234.3 224.3 241,8 |
* Грозненская парафинистая нефть.
** Взяты константы скорости первой стадии реакции. •** Ромашкннская нефть.
![]()
(18)
После интегрирования от Т\ до Т2 получаем:
где h и k2 — константы скорости при и Гг (в градусах абсолютной шкалы); R — газовая постоянная, Дж/моль; Е — энергия активации, Дж/моль.
Энергия активации определяется как избыточная (над средним уровнем) энергия, которой обладают молекулы, вступающие в реакцию. Чем выше энергия активации, тем больше энергетический барьер, который надо преодолеть для превращения обычных молекул в активные. По энергии активации можно также судить о том, насколько данная реакция или промышленный процесс чувствительны к температуре. Из уравнения (18) видно, что чем выше энергия активации, тем больше различаются константы скорости крекинга для данного температурного интервала. В среднем энергия активации при термическом крекинге составляет 210— 250 кДж/моль (50—60 ккал/моль) для реакций разложения и « 125 кДж/моль для реакций уплотнения.
Из уравнения Аррениуса следует, что в области более высоких температур, когда величина 1 /Т2—1/Т\ уменьшается (при постоянном AT=Ti—Т2), отношение констант скорости также падает, т. е. изменение температуры меньше влияет на скорость реакции.
По закону Вант-Гоффа, скорость химической реакции увеличивается в 2—4 раза при повышении температуры на каждые 10 °С. Этот закон применим для крекинга только в ограниченных температурных пределах. Число градусов, на которое необходимо повысить температуру, чтобы удвоить скорость реакции, называют температурным градиентом скорости. Обозначим его через а. Если продолжительность крекинга при температуре i0 равна то, то для той же глубины разложения продолжительность крекинга при U равна ti=to/2; при повышении температуры до t2 продолжительность крекинга станет равной T2=Ti/2, или х2=Хо/(2-2), и т. д.; при некоторой температуре tn имеем
![]()
(19)
где а — средний температурный градиент в интервале от to до tn. Это простое выражение позволяет приближенно подсчитать необходимую продолжительность крекинга при данной температуре, если известны его продолжительность при любой другой температуре и температурный градиент скорости крекинга.
Пример. Необходимое время пребывания сырья в трубах реакционного змеевика крекииг-печи 3 мин; средняя температура потока 470 °С. На сколько градусов следует повысить температуру, чтобы получить -ту же глубину разложе-
Таблица 4. Температурный коэффициент скорости* термического крекинга и температурный градиент, необходимый для удвоения скорости реакции
|
Сырье | Энергия активации, кДж/моль |
Температурный коэффициент скорости | Температурный градиент скорости, °С | ||||||
| при 400 °С | при 450 °С | при 500 °С | при 550 °С | при 400 °С | при 450 °С | при 500 °С | при 550 “С | ||
|
Газойль | 230 |
1,83 | 1,68 |
1,58 | 1,50 |
11,5 | 13,3 |
15,2 | 17,2 |
| Лнгроин | 270 |
2,04 | 1,86 |
1,72 | 1,62 |
9,7 | 11,2 | 12,8 | 14,6 |
| То же | 290 |
2,16 | 1,94 |
1,79 | 1,67 |
9,0 | 10,4 |
11,9 | 13,5 |
* Температурным коэффициентом скорости реакции называют относительное увеличение скорости прн повышении температуры иа 10 °С. .
ния за 2 мнн, если известно, что температурный градиент скорости крекинга равен а = 13 °С?
Расчет ведем по формуле:
х =2
Логарифмируя, получаем
lgl,5 = -^-1347- lg
2
откуда <яг478°С, т. е. температуру следует повысить на 8°С.
Температурные градиенты скорости различны для разных пределов температур и, как видно из уравнения Аррениуса, увеличиваются с повышением температуры. В табл. 4 приведены усредненные температурные градиенты скорости крекинга, полученные С. Н. Обрядчиковым применительно к статическим условиям.
¦ В уравнении (19) температурный градиент а взят при температуре, средней между Тх и Г2. Можно считать, что в некоторых ограниченных пределах температура и продолжительность крекинга взаимозаменяемы. Известно, например, что при «эквивалентных» по температуре и продолжительности условиях крекинга можно получать на промышленных установках практически один и тот же выход бензина с одинаковым октановым числом. Однако влияние абсолютного значения температуры на процесс крекинга несомненно.
Выше отмечалось, что умеренные температуры крекинга способствуют реакциям уплотнения; при этом энергия активации реакций уплотнения значительно меньше, чем для реакций разложения. Таким образом, чтобы увеличить выход продуктов разложения (газ, бензин) и снизить выход продуктов уплотнения (остаток, кокс), следует иметь в реакционной зоне по возможности высокую температуру при соответственной небольшой продолжительности
Таблица 5. Влияние температуры на выход продуктов разложения и уплотнения при крекинге я-цетана
Данные М. Д. Тиличеева н К. И. Зиминой
|
Температура, “С | 1 Глубина превращения н-цетана, % (Масс.) | Выход на про и-детан, продукты разложения |
реагировавший % (масс.) продукты уплотнения | Соотношение продуктов разложения н уплотнения |
| 375 | 52,2 | 55,8 | 44,2 |
1,26 |
| 400 | 53,5 | 62,2 ' |
37,8 | 1,65 |
| 425 | 50,8 ¦ |
72,4 | 27,6 |
2,62 |
процесса. В табл. 5 показан относительный выход продуктов разложения и уплотнения при крекинге я-цетана. Видно, что с увеличением температуры при практически одинаковой глубине превращения сырья преобладают реакции разложения.
Из вышесказанного можно сделать следующие практические выводы. Если целевыми продуктами термического процесса являются продукты разложения, целесообразно проводить его при высоких температурах и малом времени контакта. Наиболее яркий пример таких процессов — пиролиз с получением максимального выхода газа, богатого непредельными углеводородами. На современных трубчатых пиролизных установках температура в реакционном змеевике достигает 840—900 °С при времени контакта 0,1-0,3 с.
•V'
Эксплуатация старых промышленных установок термического крекинга, целевым продуктом которых являлся бензин, показала, что продукт уплотнения (кокс) отлагается главным образом в зоне умеренных, а не максимальных температур; этому способствует и повышенная доля жидкой фазы в сырье. Процессу коксования, целевым продуктом которого является кокс, также благоприятствуют умеренные температуры (450—470°С), способствующие реакциям уплотнения. В табл. 6 представлены данные по кинетике коксования тяжелого ароматизированного сырья, полученные на проточной установке. Обращает на себя внимание относительно малое влияние повышения температуры в реакторе на выход кокса и отсюда — невысокая энергия активации реакций уплотнения.
Если сравнивать углеводороды с одинаковым числом атомов Углерода в молекуле или узкие фракции с одинаковыми пределами выкипания, можно установить, что наименьшей термической стабильностью отличаются парафиновые углеводороды, а наибольшей— голоядерные ароматические. Нафтены занимают промежуточное положение. Например, константы скорости разложения ^'Гексана, циклогексана и бензола (углеводороды С6), вычислен-
Таблица 6. Кинетические показатели замедленного коксования крекннг-остатка тяжелого дистиллята смеси восточноукраннских нефтей при различных средней температуре н объемной скорости подачн сырья
Данные Г. Д. Голубковой и автора этой книги (плотность сырья р|О=1,0438, коксуемость 11,6%, содержание асфальтенов 7,7%)
|
При 450 | с |
При 470 | ’С |
Прн 500 °С | |||||
|
Показатели | 0,07 ч-1 |
0,10 ч—1 | 0,15 ч-1 | 0,07 ч-1 | 0,10 4-1 | 0,15 ч-l j | 0,07 ч-1 | 1 СГ о о' | I W сГ |
| Глубина превращения сырья (сумма кокса, бензина и газа), % (масс.) | 35,6 |
34,0 | 31,0 |
38,5 | 36,0 |
31,5 | 42,3 |
40,0 | 36,2 |
| Кажущаяся константа скорости k, ч-1 | 0,0080 | 0,0080 | 0,0080 | 0,0142 | 0,0142 | 0,0142 | 0,0240 | 0,0240 | 0,0240 |
| Температурный коэффициент скорости реакции |
1,26 | 1,26 |
1,26 | 1,24 |
1,24 | 1,24 |
1,22 | 1,22 | 1,22 |
| Температурный градиент скорости реакции, °С |
30,4 | 30,4 |
30,4 | 31,8 |
31,8 | 31,8 |
34,3 | 34,3 |
34,3 |
ные по уравнению мономолекулярной реакции при 500 °С, относятся друг к другу как 6,8:0,25:0,17, а для «-декана, декалина и нафталина (углеводороды Сю) — как 49: 3,7: 0,008.
В то же время в алкилароматических углеводородах алкильная группа (связь С аром Салк) отщепляется значительно легче, чем происходит разрыв ароматического кольца. Например, для деалки-лирования а-метилнафтаЛина константа скорости в 960 раз больше, чем для разрыва нафталинового кольца при той же температуре. С увеличением длины боковой цепи (т. е. когда при крекинге происходит разрыв связи Салк—Салк) константы скорости разложения резко возрастают. Если длина цепи алкилароматического углеводорода значительна, то по термической стабильности он приближается к парафиновому углеводороду с тем же числом атомов углерода. Так, константа скорости крекинга гексадецилбензола (СбНб—СюНзз) лежит между константами скорости к-цетана (н-С16Н34) и доказана (к-СггЙЦе).
Аналогичное явление наблюдается и для нафтенов с длинными боковыми цепями: они по термической стабильности приближаются к соответствующим парафинам.
Поскольку для всех парафиновых, алкилнафтеновых и алкил-ароматических углеводородов термическая стабильность с удлине-
нием цепи падает, этот же
показатель для более тяжелого по
фракционному составу сырья ниже, чем
легкого. В то же время ароматические
углеводороды (независимо от числа колец)
способны в первую очередь к реакциям
конденсации, и даже такой
высокомолекулярный углеводород, как
коронен (мол. масса 300), образующий структуру
из семи колец, перегоняется без разложения
при температурах, превышающих 500°С. Однако в
нефтяном ^ 80 г-"—¦¦-
- —
| / | |
|
,'уГ 2 &. | |
| -* | |
|
- | |
| - АмА | 1 " & |
| - 1 N | 1 ч |
Дородов. !«,"»/,
Значительный теоретический и <§ цщ m 450 m Ш
практический интерес представляет Температура°С
кинетика разложения сернистых рис и Температурлая зависи.
соединении. Данные ПО этому во- мость степени разложения сернис-
просу весьма ограничены. Ввиду ,тых соединений, содержащихся в
ТОГО ЧТО конечным продуктом разло- . хроматографических фракциях,
жения сернистых соединений нефти кинской^ефти” Гудрона ромаш'
ЯВЛЯеТСЯ сероводород, его ВЫХОД j _МОНОцНклические ароматические уг-
при крекинге сернистого сырья МО-' леводороды; 2 — бициклические арома-
« г тические; 3 — полициклические; 4 —
жет до некоторой степени служить смолы; 5 — асфальтены; 6 - исходный
показателем термической стабиль- wp°h-
ности сернистых соединений.
Исследование термической стабильности сернистых соединений, содержащихся в компонентах тяжелых нефтяных остатков — гуд-ронах типичных сернистых нефтей, показало, что наименьшая термическая стабильность свойственна сернистым соединениям, присутствующим в асфальтенах259. Разложение этих соединений начинается уже при 405—410 °С, и до 425—435 °С скорость их разложения превышает скорость разложения сернистых соединений из других компонентов гудрона (ароматические углеводороды, смолы). Кинетика разложения сернистых соединений, содержащихся в компонентах гудрона ромашкинской нефти, графически дана на рис. 14. Характерно, что при ужесточении крекинга, когда остальные сернистые компоненты начинают интенсивно разлагаться, степень разложения сернистых соединений, находящихся в асфальтенах, стабилизируется и лишь ненамного превышает 40%. Авторы
исследования объясняют это не высокой термической стабильностью этих соединений, а недостатком водорода, необходимого для образования H2S.
Основные факторы промышленных процессов термического превращения нефтяного сырья
Основными факторами термического крекинга нефтяного сырья > являются термическая стабильность сырья, температура и длительность процесса. Что касается давления, то оно влияет на результаты крекинга только при определенных условиях.
При рассмотрении этих факторов применительно к промышленным процессам следует учитывать фракционный и групповой химический состав нефтяного сырья. При родственном химическом составе с утяжелением фракционного состава сырья снижается его термическая стабильность.
Например, по данным М. Д. Тиличеева, при крекинге трех фракций грозненской парафинистой нефти, содержащей «60% парафиновых углеводородов, глубина превращения при одинаковом режиме крекинга составила 24% для бензина, 35% для керосина и 47% для солярового дистиллята.
Обогащение сырья ароматическими углеводородами значительно повышает его термическую стабильность: при пиролизе бензина, содержащего 15,4% ароматических углеводородов, газообразование было на 25% больше, чем при пиролизе того же бензина, но с добавкой 35% толуола. Высокоароматизированное сырье чрезвычайно стабильно к действию температуры: смолу пиролиза, содержащую «40% (масс.) би- и полициклических ароматических углеводородов, можно нагревать в трубчатой печи до 500—510°С без заметного разложения. Большую роль в кинетике коксоотло-жения играет относительное содержание ароматических и «-парафиновых углеводородов в. сырье: я-парафины способствуют осаждению асфальтенов из потока тяжелого сырья, нагреваемого в трубах печи, что вызывает отложение кокса на внутренних стенках труб, а ароматические углеводороды поддерживают асфальте-ны в состоянии раствора.
Сырье парафинового основания, как наименее стабильное, будет наилучшим для процессов, целевыми продуктами которых являются продукты разложения (газ пиролиза, бензиновые и газой-левые фракции установок термического крекинга под давлением). Напротив, ароматизированное сырье предпочтительно для процессов, где целевым является продукт поликонденсации — нефтяной кокс. Нафтеновое сырье имеет промежуточное значение и в зависимости от глубины крекинга может дать легкие продукты разложения типа деалкилированных нафтенов, которые, попадая в крекинг-бензин, сообщают ему более высокое октановое число, чем у бензина, полученного при крекинге парафинистого сырья. При более глубоком превращении, например при пиролизе, нафтеновое сырье дает меньше газа, но в смоле содержится больше ароматических углеводородов, чем при пиролизе парафинистого сырья. При этом реакциям разложения, как было сказано выше, способствуют высокие температуры в сочетании с малой продолжительностью процесса, а реакциям уплотнения — длительное время реакции при более умеренных температурах.
При рассмотрении основных факторов термического крекинга следует учитывать, что сырье и продукты термодеструкции могут находиться в реакционной зоне в газовой или жидкой (чаще в смешанной, жидко-паровой) фазе. Для легкого дистиллятного сырья температура процесса всегда выше температуры полного
• испарения сырья. Если применяют высокое давление, температура полного испарения сырья повышается. Однако и в этом случае сырье обычно находится в газовой фазе, так как температура в зоне реакции выше критической температуры сырья. Иное положение создается при крекинге тяжелого остаточного сырья. В этом случае, как правило, сырье и продукты находятся в смешанном состоянии (жидкость и пары): чем выше температура и чем ниже давление, тем больше доля газовой фазы. Фазовое состояние продуктов крекинга зависит и от глубины превращения сырья, так как при значительном выходе продуктов разложения высокое парциальное давление их паров обеспечит переход в газовую фазу и более высококипящих продуктов уплотнения.
Наличие факторов, способствующих переходу всей реакционной массы в газовую фазу, уменьшает опасность коксоотложений. Непосредственное образование углерода из газовой фазы и отложение его на внутренних стенках реакционного змеевика в виде так называемого пироуглерода возможно только при очень высоких температурах (850°С и выше).
При этом на реакции разложения углеводородов как на элементы, так и на радикалы решающее влияние оказывает относительная поверхность стенки, т. е. отношение этой поверхности к реакционному объему. При низких давлениях значительную роль играет обрыв цепей на стенках реактора в ходе радикально-цепного процесса крекинга. Повышение давления, естественно, влияет только на реакции, протекающие в газовой фазе. До определенного предела давление способствует контакту молекул и тем активизирует их взаимодействие. При дальнейшем повышении давления подвижность молекул затрудняется, и газовая фаза по свойствам все более приближается к жидкости, где радикалы окружены «клеткой» из соседних молекул («клеточный» эффект), что затрудняет развитие цепи.
Таким образом, для газофазного процесса большое значение имеет абсолютная величина давления. М. Г. Гоникберг и В. В. Воеводский показали, что при невысоких давлениях (несколько десятых МПа) его повышение увеличивает константу скорости крекинга, а при очень высоких давлениях (десятки МПа) наблюдается обратное явление. По данным А. И. Динцеса, в процессе термического крекинга н-бутана при 575°С и глубине разложения 9—13% повышение избыточного давления с 0,39 до 1,08 МПа увеличивает константу скорости с 0,007 до 0,022 с-1, т. е. примерно втрое. Г- М. Панченков и В. Я- Баранов, подвергая фракцию 300—480 °С грозненской парафинистой нефти крекингу при 510 °С и избыточном давлении 1 и 5 МПа, установили, что максимальная константа скорости соответствует давлению 1 МПа; при дальнейшем повышении давления скорость разложения снижается.
Повышение давления влияет не только на скорость реакций, но и на их направление, т. е. на состав продуктов крекинга. С увеличением давления возрастает скорость вторичных превращений продуктов разложения (полимеризация, алкилирование, перераспределение водорода). Таким образом, с повышением давления уменьшается выход газообразных продуктов распада и увеличивается количество продуктов уплотнения. Это подтверждается составом продуктов парофазного крекинга (крекинг при низком давлении) и термического крекинга под давлением.
Ниже приведен типичный материальный баланс крекинга однотипного дистиллятного сырья (керосин с относительной плотностью; 0,820) при низком и высоком давлении; в данном случае газооб-; разованию способствует не только низкое давление, но и высокая температура: 260
Данные С. Н. Обрядчикова |
| Показатели |
Парофазный | Крекниг пед |
| крекинг |
. давлением | |
|
550—560 | 500—510 | |
| Избыточное давление, МПа . Материальный баланс, % (масс.) |
0,2—0,5 | 4—5 |
| газ ........ | 32,0—32,3 | 15,0 |
|
бензиновый дистиллят |
58,5—62,2 | 75,0 |
| крекинг-остаток..... |
10,4-13,1 | 10,0 |
сырье подвергать крекингу при низком давлении, удельный объем паров и скорость их движения будут очень велики, и для их выдерживания в реакционной зоне в течение времени, необходимого для химического превращения, потребуется значительный объем реакционного пространства. Стремление к сокращению реакционного объема привело к широкому распространению крекинг-установок при повышенном давлении.
Установки крекинга, на которых перерабатывали облегченное сырье при низком давлении (установки парофазного крекинга), имели низкую производительность и, следовательно, были нерентабельными, поэтому их широкое внедрение в нефтеперерабатывающую промышленность затормозилось. Однако для такой разновидности термического крекинга, как пиролиз, где низкое давление оправдано большим выходом газообразных целевых продуктов (этилен, пропилен), а также для так называемых термоконтактных процессов низкое давление может являться положительным фактором, так как оно способствует реакциям распада и быстрому удалению из реакционной зоны продуктов первичного разложения исходного сырья.
Процессам термического крекинга, протекающим в жидкой фазе, соответствует тяжелое сырье — нефтяные остатки, тяжелые дистилляты. Если предусмотрено неглубокое разложение сырья (например,' для снижения вязкости остатка в процессе висбрекинга), конечный продукт содержит небольшое количество легких фракций (газ, бензин), которые находятся в газовой фазе. Основная масса продукта, как и исходное сырье, остается в жидкости. При наличии глубокого превращения, как это происходит в процессе коксования, крекинг протекает в камере или на поверхности теплоносителя с образованием твердого остатка и паров продуктов разложения. В процессе висбрекинга роль давления невелика — повышенное давление лишь немного увеличивает пропускную способность установки. При коксовании роль давления больше (особенно при переработке дистиллятного сырья), поскольку реакции уплотнения будут протекать не только в жидкой фазе, но и за счет конденсации паров высокоароматизированных продуктов разложения.
Общие свойства продуктов термического крекинга
В результате любого термического процесса переработки нефтяного сырья образуются газообразные и жидкие продукты; глубокие формы крекинга сопровождаются появлением твердых продуктов уплотнения (кокс).
По появлению газообразных продуктов можно судить о начале процесса, а по выходу газа при крекинге данного сырья при разных режимах можно приближенно оценить общую глубину превращения сырья (в определенных границах параметров).
Состав углеводородных газов крекинга в основном зависит от режима процесса— температуры, времени, давления. Что же касается качества сырья, оно может оказать значительное влияние только в некоторых специфических случаях. Например, пиролиз этана дает газ, весьма богатый этиленом, т. е. в основном протекает дегидрирование этана; присутствующие более тяжелые газообразные углеводороды являются уже продуктом вторичных реакций, поэтому выход пропилена и бутиленов при пиролизе этана незначителен.
Второй пример влияния состава сырья — крекинг голоядерных ароматических углеводородов; получаемый газ обогащен водородом, выделяющимся при конденсации бензола в дифенил, нафталина в динафтил и т. д.
При крекинге под давлением и при коксовании жидкого сырья (газойли, мазуты, гудроны) состав газов довольно сходен и характеризуется значительным содержанием «сухой» части (метан, этан) и умеренным (25—30%) содержанием непредельных. Такой состав обусловлен радикально-цепным механизмом процесса и нестабильностью радикалов »СзН7 и выше.
С повышением давления в реакционном объеме снижаются выход газа и содержание в нем непредельных углеводородов за счет протекания полимеризации и гидрирования. Сочетание высокой температуры и малой продолжительности процесса направлено на максимальный выход непредельных компонентов газа (этилен, пропилен и др.)-
Жидким продуктам крекинга свойственно присутствие непредельных и ароматических углеводородов. При средней глубине процесса крекинг-бензины обладают невысоким октановым числом (60—65); с углублением процесса концентрация ароматических углеводородов возрастает, поэтому октановое число повышается: бензин, получаемый термическим риформингом лигроина, имеет октановое число 70—72, а бензин, выделенный из смолы пиролиза, имеет октановое число 80 и выше. Йодные числа типичных бензинов, образующихся при термическом крекинге под давлением и коксовании, довольно высоки (80—100 г Ь на 100 г).
С утяжелением фракционного состава продуктов крекинга их непредельность снижается; крекинг-газойли, выкипающие в пределах 200—350 °С и часто используемые (после очистки) как компоненты дизельного топлива, имеют йодное число 40—50 г Ь на 100 г. Более тяжелые фракции обычно возвращают на рециркуляцию или выводят в виде тяжелого газойля (коксование) или крекинг-остатка (крекинг под давлением). В зависимости от режима процесса и качества сырья эти продукты более или менее ароматизированы. Крекинг-остатки содержат довольно много смолисто-ас-фальтеновых веществ и некоторое количество твердых частиц — карбоидов. Тяжелая часть смолы пиролиза представляет собой концентрат полициклических ароматических углеводородов; в ней содержатся также смолы, асфальтены и кар бонды.
Твердые продукты уплотнения (кокс) получаются в балансовых количествах только при одном из рассматриваемых процессов — коксовании. Кокс представляет собой продукт глубокого уплотнения полициклических ароматических углеводородов, смол и асфальтенов и содержит 94—95% углерода. Помимо углерода и водорода в зависимости от качества сырья коксования в состав кокса могут входить сера, азот, кислород, металлы.
Более подробно свойства продуктов, получаемых при термических процессах переработки нефтяного сырья, описаны в соответствующих разделах гл. IV.
ПРОМЫШЛЕННЫЕ ПРОЦЕССЫ ТЕРМИЧЕСКОГО КРЕКИНГА
ТЕРМИЧЕСКИЙ КРЕКИНГ ПОД ДАВЛЕНИЕМ
Наиболее распространенной формой промышленного процесса термического крекинга до 40—50-х годов был крекинг при высоком давлении. В 1935 г. был разработан проект отечественной двух* печной крекинг-установки системы «Нефтепроект». В одной печи подвергали легкому крекингу тяжелую часть мазута, а во второй осуществляли глубокий крекинг керосино-газойлевых фракций, содержащихся в исходном мазута и образовавшихся после легкого крекинга. Таким образом был соблюден необходимый при крекинге принцип селективности (избирательности): тяжелую часть сырья (выкипающую выше 350 °С) подвергали крекингу при мягком температурном режиме (470—480°С), а керосино-газойлевые фракции (200—350 °С) —при более жестком (500—510 °С).
В 40—50-х годах на зарубежных н отечественных заводах существовали установки термического крекинга следующих типов:
однопечные — для крекинга газойлевых н соляровых фракций;
то же — для глубокого крекинга (термический риформииг) лигроииовых фракций;
двухпечные — в основном для крекинга мазутов;
многопечные (с тремя и четырьмя печамн) — для крекинга полумазутов и мазутов.
На однопечных установках крекинга соляровых фракций подвергали сырье крекингу с рециркуляцией; прн этом за однократный пропуск получали 15—25% бензина, а общий выход бензина составлял 50—60%. Температура на выходе из-реакционного змеевнка равна »500°С, давление на входе в печь 4—5 МПа. С увеличением спроса на дизельное топливо газойлево-соляровые фракции стали дефицитными, и установки для крекинга соляровых фракций перестал» строить.
Исходным сырьем термического риформинга (стр. 15) были низкооктановые лигроиновые (реже керосиновые) фракции. Таким образом, фракционный состав сырья и получаемого крекинг-бензи* ка частично совпадал, что указывало на необходимость глубокого преобразования молекул исходного сырья для получения из них ароматизированных бензинов с удовлетворительным октановым числом. Действительно, октановые числа риформинг-бензинов (в среднем 70—72) наиболее высоки по сравнению с октановыми числами бензинов других видов термического крекинга под давлением (60—65 для бензинов, получаемых крекингом мазута). Температурный режим термического риформинга жесткий и зависит от фракционного состава сырья: для бензино-лигроиновых фракций температура риформинга достигает 550—560 °С при 5—6 МПа.
Октановое число получаемого бензина возрастает с увеличением? глубины превращения. В настоящее время доля термического риформинга в нефтеперерабатывающей промышленности невелика,, так как широкое промышленное развитие получил более эффективный каталитический риформинг.
Наибольшее распространение среди процессов термического1 крекинга под давлением получил крекинг мазутов по двухпечной схеме. Поточная схема крекинга такова:
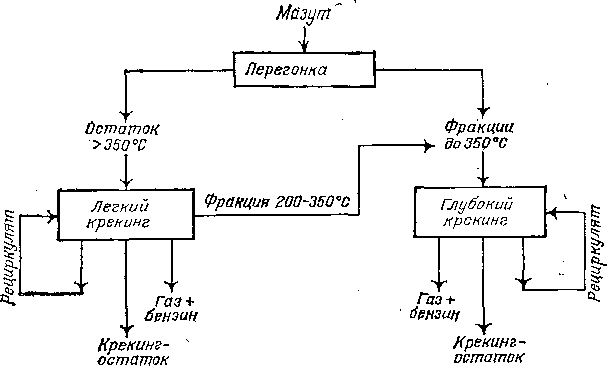
Из схемы видно, что в печи глубокого крекинга в качестве свежего сырья используют керосино-газойлевые фракции прямогонного происхождения и фракцию 200—350 °С, полученную легким крекингом остатка. При утяжелении исходного сырья единственным источником питания этой печи является газойлевая фракция легкого крекинга, выход которой незначителен, и, в конечном счете, тяжелое остаточное сырье оказывается целесообразным перерабатывать на однопечной установке.
Характерно, например, что если на первых установках системы «Нефтепроект» соотношение загрузки в печах тяжелого и легкого сырья было равно 1,5 : 1, то строившиеся с 50-х годов установки системы «Гипронефтезаводы» имели печи с соответствующим отношением 4:1, причем при их эксплуатации на гудроне оказалось, что в этом случае печь легкого сырья была недогруженной.
Стремление повысить селективность крекинга привело к созданию многопечных установок: с тремя, четырьмя и даже пятью печами. В результате крекинга по многопечной схеме глубина отбора бензина практически не увеличивалась. Кроме того, несмотря на некоторые экономические преимущества (пониженные эксплуатационные затраты) многопечные установки были чрезвычайно сложны в эксплуатации, поэтому широкого развития они не получили.
Типовая технологическая схема установок двухпечного крекинга с выносной реакционной камерой. Выше сказано, что наиболее типичным сырьем современных крекинг-установок являются полу-гудроны и гудроны. При этом крекинг'бензин становится побочным продуктом, а выход целевого крекинг-остатка составляет 80—90% на сырье. В этом случае в технологической схеме может быть предусмотрена только одна печь. Однако типовые установки системы «Гипронефтезаводы», спроектированные к началу 50-х годов, были рассчитаны и на переработку остатков, более легких по фракционному составу; исходя из этого была принята схема двухпечного типа (рис. 15).
Сырье (мазут) подают насосом Н-1 через теплообменники Т-1; часть сырья (~7з) после теплообменников поступает в нижнюю часть колонны К-3, а остальное — в испаритель К-4 низкого давления. Разделение сырья на два потока позволяет полнее использовать избыточное тепло паров в аппаратах К-3 и К-4. Поток сырья, обогащенный тяжелыми газойлевыми фракциями в испарителе К-4, также направляют в колонну К-3, с низа которой горячим насосом Н-2 качают смесь сырья и тяжелого рециркулята в печь П-1. Газойлевые фракции со сборной тарелки колонны К-3 вторым горячим насосом Н-3 подают на глубокий крекинг в печь П-2.
Продукты легкого и глубокого крекинга из обеих печей поступают для углубления крекинга в верхнюю часть выносной реакционной камеры К-1. Крекинг-остаток отделяется в испарителе К-2 высокого давления. Давление в аппарате К-1 («?2 МПа) снижают до давления в испарителе К-2 (1 МПа) посредством редукционного вентиля. Крекинг-остаток из испарителя К-2 самотеком перетекает в испаритель К-4 низкого давления, где из крекинг-остатка выделяются пары газойлевых фракций, вступающих в контакт с потоком исходного сырья. Некоторое количество несконденсиро-вавшихся фракций уходит с верха испарителя К-4, проходит конденсатор-холодильник Т-2 и из газосепаратора Е-2 низкого давления идет в мерник, а также используется в виде циркулирующего потока, орошающего колонну К-4. Поток паров из испарителя К-2 идет на разделение в колонну К-3.
Наиболее тяжелые фракции конденсируются и служат рецир-кулятом для печи П-1 тяжелого сырья. Конденсат легких газойлевых фракций отбирают со сборной тарелки колонны К-3. С верха этой колонны уходят пары бензинового дистиллята и газ; они конденсируются в конденсаторе-холодильнике Т-3, доохлаждаются в холодильниках Т-5 и разделяются в газосепараторе Е-1 высокого давления. Газ направляют на абсорбционно-газофракционирую-
я x ^ 05=: sp®
Sf X 2 « <u et
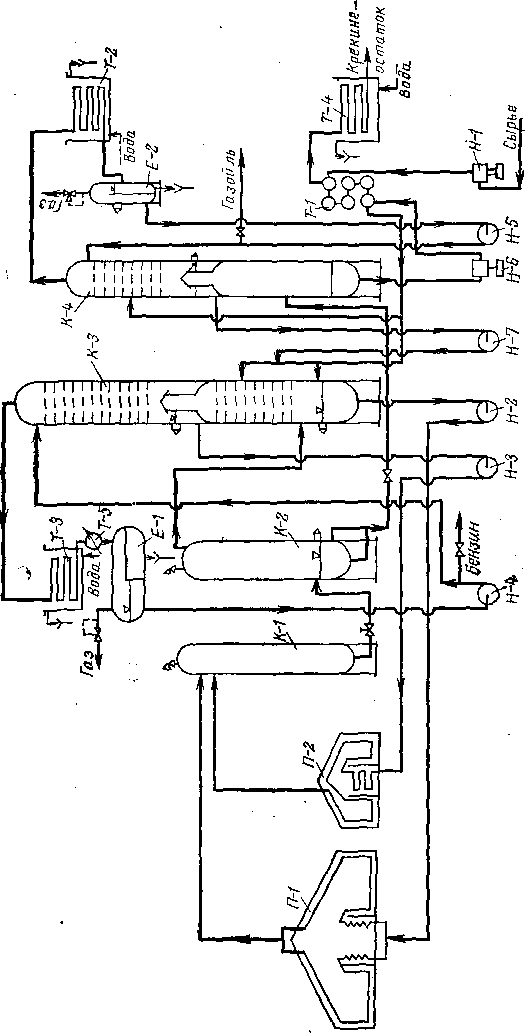
те 35
S.’gg
о о
~ ч * 2 = 2 w (U X gH =
§ 1 a S'? ° ® со
1 «-§• *7 я S
i *ss
) F> CO
TO »* t_ Pi I
К ?cn
S IK PS я
о л я
^ Д щ
О ^ * о (Q
О Н я
"I
"g?
О с'Т з 2 ~*<в
Э ? « ?
о с о :
° « * ?
F'*'» »д _ I a ' ST <5^ О h i> а о
Я С 3 .- «5
S> ev ? = I
&ci?5^
о г^-, 8
« |S I -
* . 1 4;
йй
=Г
CJ
С
X
>»
со
t=t
{-
л « Я ? «5 ^ О
о
? о5' 3 *8? I
OS'* JS О Г
К
Он
S-1S
ь .Otf я М я
? °- I 1
В" a; I 1
с
I
т ^2* S
fc; аз*-« ай
щую установку (АГФУ), а бензин подают на стабилизацию. Отгон стабилизации также направляют на газофракционирование.
Ниже приводятся основные технологические показатели установки двухпечного крекинга:
Температура.
°С
470—490
530—545
500
450—470
430—440
390—410
280—320
210—220
400—415
Давление, МПа
2,2-
2,2-
¦2,7
-2,8
| 0,85—1,2 0,85—1,2
, 0,8-1,2 0,15—0,3
Аппарат
П-1 (выход.....
П-2 (выход) . . . ...
К-1
верх......
низ.......
К-2 . .......
К-3
низ......
аккумулятор легкого сырья
верх......
К-4 (низ)......
Печи крекинга тяжелого сырья несколько различаются: печь П-1, рассчитанная на большую пропускную способность, двухпоточная, а печь П-2 однопоточная. В обеих печах реакционные змеевики размещены в конечной (по ходу сырья) части радиант-ных труб. Реакционная камера К-1 работает с низким уровнем жидкости (стр. 29), что обеспечивает преимущественное заполнение ее парами продуктов крекинга.
Ниже дан примерный материальный баланс (в % масс.) установок двухпечного крекинга при переработке мазута (остаток >350 °С сернистой нефти типа ромашкинской) и гудрона (остаток >460 °С высокосернистой арланской нефти):
|
Мазут | Гудрон |
| 66,3 | 86,8 |
| 5,3 | _ |
| 19,7 | 6,7 |
| 3,6 | 3,0 |
| 3,5 |
2,3 |
| 1,6 |
1,2 |
Поток
Крекииг-остаток Крекинг-гайзоль (фракция 200—350 °С) . .
Крекинг-бензин .
Отгон стабилизации Крекинг-газ Потери ....
Требуемая вязкость крекинг-остатка зависит от марки получаемого котельного топлива и составляет примерно ВУ8о=15° (для топочного мазута 100); товарное котельное топливо можно получать компаундированием крекинг-остатка с тяжелыми газойлями вторичного происхождения.
В связи с тенденцией к углублению переработки нефти переработка гудронов методом термического крекинга оказалась нерациональной. Сернистые гудроны после легкого термического крекинга дают котельное топливо с не меньшим содержанием серы, чем в исходном гудроне. Сжигание такого топлива без смешения его с менее сернистым недопустимо, так как сопровождается от-
равлением атмосферы сернистым ангидридом. Однако на некоторых установках (даже позднего типа) висбрекинг еще используют, но осуществляют его по однопечной схеме.
Установки висбрекинга гудрона входят в состав отечественных комбинированных установок ГК-3/1. Горячий гудрон с низа вакуумной колонны поступает в печь висбрекинга и проходит ее двумя потоками. Реакционной камеры нет, продукты крекинга поступают непосредственно в эвапоратор, с низа которого выводят крекинг-остаток, а газовую фазу направляют в колонну. С верха колонны выводят пары бензина и газ, а сбоку через отпарную колонну— дизельную фракцию. После конденсатора жирный газ отделяют в газосепараторе от нестабильного бензина, который идет далее на облагораживание в блок каталитического крекинга (его закачивают в линию подачи вакуум-газойля). Остаток с низа колонны возвращают на рециркуляцию в печь висбрекинга. Выходы продуктов висбрекинга при переработке нефти типа ро-машкинской таковы: ?^80% (на гудрон) крекинг-остатка (котельное топливо), ~8% дизельной фракции, остальные 12%—газ и бензин. Для прекращения реакций крекинга в линию паров из эвапоратора предусмотрена подача охлаждающей струи (квен-чинг). Как ясно из описания схемы, блок висбрекинга содержит всего одну печь, т. е. схема достаточно проста и компактна.
Некоторые установки термического двухпечного крекинга используют для других целей, например для получения сажевого сырья. Сажа находит широкое применение в, народном хозяйстве, главным образом как наполнитель резин, а также в лакокрасочной, полиграфической и других отраслях промышленности. В качестве сырья для производства сажи используют высокоаромати-зированные дистилляты, содержащие би- и полициклические ароматические углеводороды; в качестве исходных дистиллятов применяют, в частности, газойли каталитического крекинга или их смеси с экстрактами от селективной очистки масел.
Для дальнейшего облагораживания этого сырья (разложение алкилароматических углеводородов — отрыв боковых цепей, разложение парафиновых углеводородов) его подвергают термическому крекингу по обычной схеме: сырье разделяется в колонне К-3 на легкую и тяжелую части, которые после смешения соответственно с легким и тяжелым рециркулятом подают в печи П-2 и П-1. Крекинг в обеих печах отличается более жестким режимом, чем при запроектированном процессе крекинга остаточного сырья. Так, на выходе из печи П-1 температура достигает 500 °С (вместо 470— 490°С), а на выходе из печи П-2 — 550°С (вместо 530—545°С). Это объясняется высокой термической стабильностью исходного аРоматизированного сырья. Для увеличения выхода целевого продукта (термогазойль) в дополнительном испарителе К-4 давление снижено до 0,1 МПа. Было предложено также, чтобы еще увеличить отбор термогазойля, дополнить схему вакуумным испарителем. В этом случае выход целевого продукта превышает 50% на сырье (остальное — газ, бензин и крекинг-остаток).
Термогазойль, получаемый из сернистого сырья, имеет следующие примерные показатели: относительная плотность 0,993;
по = 1,5770; 3,27% серы, 75,5% ароматических углеводородов
(в том числе 70,3% би- и полициклических); коксуемость 0,84%;
индекс корреляции ИК=90,2.
Индекс корреляции является важным показателем сажевого сырья. Он выражает зависимость между плотностью и температурой кипения для углеводородов различных рядов. Для расчета индекса корреляции выведена формула
48 640
ИК= 473,7р1° —456,8 +—f— (20)
где р!0—относительная плотность сырья, а Т — средняя температура кипения
нефтепродукта, К. Для облегчения подсчетов по уравнению (20) составлена номограмма261. Согласно техническим нормам на сажевое сырье, индекс корреля-
ции не должен быть менее 90.
Установки термического крекинга используют также для увеличения сырьевых ресурсов для некоторых каталитических процессов — каталитического крекинга, гидрокрекинга. Например, в г. Грозном (ГрозНИИ и . Гипрогрознефть) 'был предложен процесс деструктивной вакуумной перегонки (ДВП). Согласно схеме (рис. 16), мазут проходит через печь 1 и подвергается там легкому крекингу. В колонну 2 для увеличения доли отгона подают водяной пар. Отгон ,направляют непосредственно в реактор установки каталитического крекинга, а остаток поступает в вакуумную колонну 3, где за счет перепада давления происходит дополнительное испарение фракций, также направляемых на 'каталитический крекинг.
На.
каталитический
йигг
Газы
разложения
Конденсат \/Нренинд - остаток
Мазут
Рис. 16. Схема деструктивной вакуумной перегонки (ДВП):
1 — печь; 2 — колонна (избыточное давление 0,2 МПа); 3 — вакуумная колонна; 4 — емкость; 5 — эжектор.
По данным ГрозНИИ, двухступенчатая деструктивная вакуумная перегонка -сернистого мазута позволяет на 25—30% уменьшить выход остатка >500 °С и на 39—52% увеличить общий выход дистиллятов. Подобное сокращение выхода остатка целесообразно, так как преобладающая доля каталитических ядов (металлы, смолы, азотистые соединения, сера) содержится в остаточном сырье (особенно из сернистых нефтей), а дистиллятное сырье, даже содержащее продукты термического разложения, значительно легче подвергать последующей каталитической переработке.
Следует упомянуть о чисто нефтехимических вариантах использования термического крекинга. Известен процесс термического крекинга парафинов с целью получения а-олефинов — сырья для производства моющих средств, поверхностно-активных веществ, спиртов и др. Процесс протекает в газовой фазе, при 550 °С и небольшой продолжительности, обусловленной малой глубиной разложения сырья. Можно упомянуть также процесс термического гидродеалкилирования алкилароматических углеводородов, входящих в состав экстрактов каталитических газойлей и катализатов риформинга. Целью этого процесса является получение нафталина или бензола262.
КОКСОВАНИЕ ТЯЖЕЛОГО НЕФТЯНОГО СЫРЬЯ
Глубина термического крекинга тяжелых нефтяных остатков ограничена образованием кокса. При переработке особо тяжелого сырья на установках висбрекинга конечными продуктами являются только газ, бензин и крекинг-остаток, в котором приходится оставлять все газойлевые фракции, чтобы получить котельное топливо стандартной вязкости, т. е. глубина крекинга весьма невелика.
Выход светлых заметно увеличивается, если термический крекинг тяжелого сырья вести с высоким выходом кокса, в котором концентрируется значительная часть углерода исходного сырья, а выход продуктов разложения (фракции газойля, бензин, газ) возрастает.
Например, при коксовании гудрона сернистой .нефти (в каме: рах) при выходе кокса 24% образуется 16% бензина (до 205°С), 26% керосино-газойлевой фракции (205—350°С) и 23% тяжелого газойля (>350°С). Все эти дистилляты содержат непредельные углеводороды, т. е. нестабильны; если перерабатывают сернистое сырье, то эти дистилляты к тому же и сернистые, т. е. нуждаются в облагораживании. Бензин имеет невысокое октановое число, но он может быть подвергнут гидроочистке с последующим каталитическим риформингом и дает л?80% масс. (т. е. 16-0,8=12,8% масс. на сырье коксования) высококачественного бензина с октановым числом не ниже 80. Керосино-газойлевую фракцию после гидро-очистки для удаления сернистых соединений и непредельных углеводородов используют как компонент дизельного топлива. Выход последнего при гидроочистке составляет я^95% масс. (т. е. 26-0,95 = 24,7% масс, на сырье). Наконец, тяжелый газойль может служить компонентом сырья каталитического крекинга или гидро-крекинга. При каталитическом крекинге получается не менее 40— 45% (масс.) бензина и 20% (масс.) легкого газойля, т.е. суммарно 60—65% (масс.) светлых (или 23-0,60 = 13,8% на сырье коксования). В итоге продукты коксования после переработки или облагораживания дадут дополнительный выход светлых:
12,8 + 24,7-)- 13,8 = 51,3% (масс.) считая на сырье коксования
Если гудрон составляет 30% от нефти, то соответствующий дополнительный выход светлых на нефть равен: 51,3-0,3=15,4%. Таким образом, коксование служит одним из путей углубления переработки нефти.
Еще больше распространен процесс коксования для получения нефтяного кокса. В этом случае предпочтительно подвергать коксованию малосернистое сырье, так как содержание серы в коксе нормировано.
Потребность в нефтяном коксе, как более дешевом и высококачественном материале, чем кокс, получаемый на основе угля (так называемый пековый), весьма значительна и непрерывно возрастает. Основной потребитель кокса — алюминиевая промышленность: кокс служит восстановителем (анодная масса) при выплавке алюминия из алюминиевых руд. Удельный расход кокса на производство алюминия весьма значителен и составляет 550— 600 кг на 1 т. Из других областей применения кокса следует назвать использование его в качестве сырья для изготовления графи-тированных электродов для сталеплавильных печей, для получения карбидов (кальция, кремния) и сероуглерода. Специальные сорта кокса применяют как конструкционный материал для изготовления химической аппаратуры, работающей в условиях агрессивных сред.
Промышленный процесс коксования осуществляют на установках трех типов:
1) периодические, в коксовых кубах;
2) полунепрерывные, в необогреваемых коксовых камерах;
3) непрерывные, в псевдоожиженном слое кокса-теплоносителя.
Периодическое коксование в кубах |
Как и всякий периодический процесс, коксование в кубах мал<1 производительно и, следовательно, неэкономично. Однако этол процесс еще применяют, обычно в тех случаях, когда ресурсы! сырья относительно невелики, например при коксовании смолы пиролиза, направленного на получение не олефинсодержащего гаЧ за, а ароматизированной смолы. I
Коксовый .куб представляет собой цилиндрический гори зон-] тальный аппарат диаметром 2—4,5 м и длиной 10—13 м. Сырья загружают в куб и постепенно нагревают, подавая топливо через форсунку, расположенную в топке под кубом. Примерно при 300 °С начинают выделяться пары, которые уходят через шлемо-вую линию и поступают в систему конденсации и охлаждения. По мере нагревания куба интенсивность выделения погонов усиливается, достигая максимума при 360—400 °С в газовой фазе. Обычно максимальная температура паров 450°С, после чего она снижается вследствие прекращения выделения погонов.
Для завершения процесса образующий на дне куба коксовый «пирог» прокаливают в течение 2—3 ч. По окончании прокалки постепенно снижают температуру в топке, гасят форсунку и охлаждают куб, вначале подавая водяной пар (который одновременно удаляет из куба газообразные углеводороды и пары), а затем воздух (после охлаждения куба до »300°С). Из охлажденного куба выгружают кокс через разгрузочный люк,- Выгрузка кокса почти не механизирована и продолжается от 2 до 4 ч.
Для периодического коксования характерно, что продукты разложения непрерывно удаляются из реакционной зоны, а остаток в кубе все более утяжеляется и постепенно превращается в кокс. Достоинством получаемого кокса является низкое содержание летучих, поэтому не требуется дополнительных прокалочных устройств.
В настоящее время этот процесс применяют в основном для коксования смолы пиролиза при получении кокса специальных видов — электродного и конструкционного. В обоих случаях подвергают коксованию высокоароматизированную тяжелую смолу, получаемую пиролизом керосиновых или газойлевых фракций. В состав этой смолы входят в основном полициклические ароматические углеводороды, смолы и асфальтены; в ней имеется и ‘некоторое количество карбоидов. Например, образец пиролизной смолы, предназначенный для получения электродного кокса, имел плотность 1218,5 кг/м3 и содержал 1,9% (масс.) карбоидов, 3,4% (масс.) карбенов, 16,4% (масс.) асфальтенов, 2,5% (масс.) смол и 75,8% (масс.) масел. Масла состояли на 34% из фенантренов и антраценов и содержали другие полициклические ароматические углеводороды. Однако в связи с увеличением потребности в коксе пиролизного происхождения пиролизные смолы тоже начали перерабатывать на более совершенных установках полунепрерывного (замедленного) коксования.
Исследованием периодического коксования занимались А. Ф. Красюков, С. Н. Обрядчиков и М. X. Левинтер. Определение состава жидких продуктов коксования в процессе превращения сырья в кокс показало, что масла и смолы, содержащиеся в сырье, постепенно переходят в асфальтены, которые превращаются в карбоиды. Конечные продукты коксования — карбоиды и термически стабильные ароматизированные масла.
Периодическое коксование дает наибольший выход кокса по сравнению с другими способами. Так, при периодическом коксовании битума (плотность 1019 кг/м3) выход кокса доходит до 30%, а при полунепрерывном всего до 21,0%. Естественно, что большему выходу кокса соответствует меньший выход дистиллята, имеющего при этом более легкий фракционный состав. Отмеченные особенности периодического коксования объясняются тем, что процесс протекает при относительно низких температурах, что замедляет удаление продуктов разложения из реакционной зоны и благоприятствует реакциям уплотнения.
Полунепрерывное коксование в необогреваемых коксовых камерах
Этот промышленный процесс коксования получил наибольшее распространение как в Советском Союзе, так и за рубежом: кокс получается в виде кускового, и сортировка его по размерам позволяет легко выбрать фракцию (обычно 25 мм и выше), пригодную для последующей прокалки в печах существующих конструкций. Схема установки достаточно проста; в ней предусмотрена рециркуляция тяжелой части жидких продуктов. Выход кокса выше, чем при непрерывном процессе. Выгрузка кокса полностью механизирована. Мощность установок достигает 1,5 млн. т в год по сырью и соответствует масштабам современных нефтеперерабатывающих заводов. В Советском Союзе проектируются и находятся в эксплуатации установки подобного типа, мощностью 300, 600 и 1500 тыс. т сырья в год.
Технологическая схема установки. На рис. 17 представлена схема установки пропускной способностью 600 тыс. т в год, рассчитанная на переработку малосернистого сырья. Установка имеет четыре коксовых камеры и две трубчатых нагревательных печи.
Исходное сырье насосами 5 подают двумя параллельными потоками в трубы подовых и потолочных экранов печей 3 к 4, нагревают там до 350—380 °С и направляют в нижнюю часть ректификационной колонны 6. В этой секции сырье встречается с пото ком парообразных продуктов коксования из двух параллельно ра ботающих камер 1. В результате этого контакта наиболее тяжела часть паров конденсируется и смешивается с сырьем; в нижне" части колонны образуется таким образом смесь сырья с рецирку лятом, обычно называемая вторичным сырьем. Если сырье содержало некоторое количество легких фракций, они в результате контакта с парами из камер испаряются и уходят в верхнюю часть колонны 6.
Вторичное сырье с низа колонны 6 насосами 7 возвращают в печи 3 >и 4 — в верхнюю часть конвекционных труб и правые подовые и потолочные экраны. Эта часть труб относится к «реакцион-
. л л , ь t*
? о о 7 * *
2 S ^ а> а>
Я-,еч
*
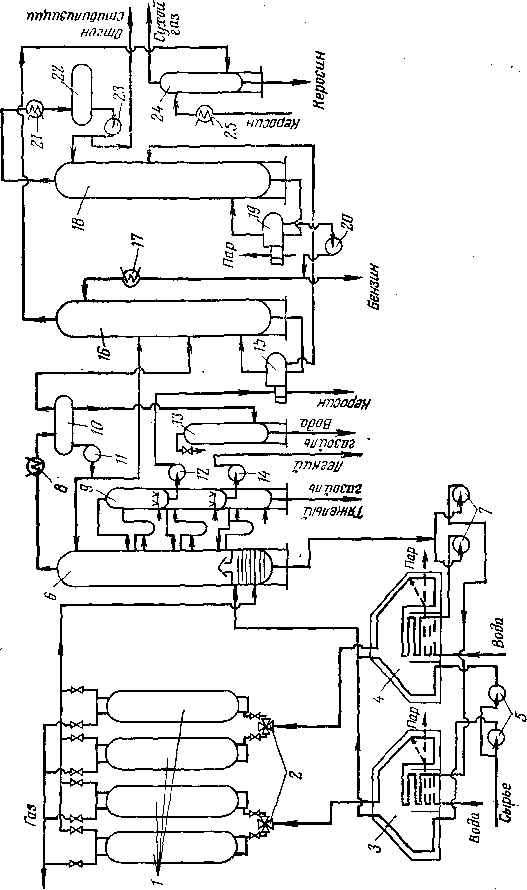
2 3 л
0 О X
So# * о о
В! S § S I *
8*3 w Я
^«УЧ-*
со 2? О
s . tS '8‘?> я « *N rt {«. *55 tt** е? 4>-, 5!
СХ \о
1 5'н <е
3 ..2
у J3
° ч *
® S s
Й ч к л 5i j и ^ ч
ss§
eg§
S?§
j O X
I X , 45 2 * ... «с Ё 1 ^ .. ё^с;
R 1 ..
S 'So
sa > я & eo «ID О »=?§¦
О (j
\o
•Q os V
53'
• S*
“ 6.S*
о ^ я cf U <D С Я
S 3°?
X CQ ^
S o|b O) !=c 1 ? e; o^5
Э * -g
S * S tr
4 s Д к ~ н S 21
(D _, *0*
53 ег F>
5 I .
o^o а,
СО
О Сц q 33 о W
g gilf У E^SiO
^ S ¦*
И fi •-
О СС ° !=
Я '-'2 з
#s о 33 Я
о Б :
« Et
<D
5 P О
* i °- я m %
= I =
ii.s'§
“eo‘24
S !=C ra • - о
M j to1» О 'N
h- 0 2 О = s XXO E я c « « fi ^ S „ —
s I S
Ри P-
5 a
Ct о
ному» змеевику, вторичное сырье «агревается там до 490—510 °С. Во избежание закоксовывания труб этой секции в трубы потолочного экрана подают перегретый водяной пар-турбулизатор (~3°/о на вторичное сырье), который увеличивает скорость прохождения потока через реакционный змеевик. Паро-жидкостная смесь вводится параллельными потоками через четырехходовые краны «в две работающие камеры 1 (остальные две камеры в этот период подготавливают к рабочей части цикла). Входя в низ камер, горячее сырье постепенно заполняет их; так как объем камер большой, время пребывания сырья в них также значительно, и там происходит крекинг сырья. Пары продуктов разложения непрерывно уходят из камер в колонну, а утяжеленный остаток задерживается в камере.
Практика эксплуатации установок замедленного коксования показала, что процесс протекает постадийно263. Вначале тепло затрачивается на прогрев камер и испарение образующегося конденсата, что замедляет разложение. В этот период вследствие преобладания испарения над крекингом образуются дистилляты, более тяжелые по фракционному составу. Продолжительность первого периода тем меньше, чем тяжелее и смолистее сырье и чем выше температура его подогрева в печи. Так, для полугудрона первый период при 475 °С длится 8—9 ч, а при 500—510°С всего 5,4 ч; для крекинг-остатков, богатых асфальтенами, он составляет соответственно 5 и 2 ч. Именно в этот период наблюдаются «перебросы» сырья в колонну, так как уровень в камере возрастает, а постепенное повышение концентрации асфальтенов в жидком содержимом камеры вызывает вспучивание асфальтенов образующимися газами. В результате постепенного накопления коксообразующих веществ в жидком остатке он превращается в кокс.
Вторая стадия коксования сопровождается равномерным нарастанием коксового слоя и постоянными (в течение некоторого времени) выходом и качеством продуктов разложения. По мере заполнения камеры коксом свободный реакционный объем уменьшается и одновременно увеличивается средняя температура коксования; при этом качество дистиллятов снова может колебаться, а коксовый слой получается более плотным и с меньшим содержанием летучих.
Из сказанного следует, что чем выше температура нагрева сырья в печи, тем меньше опасность «переброса» остатка из камеры в колонну и тем лучше качество получаемого кокса (вследствие снижения в нем летучих). Процессы поликонденсации, свойственные коксованию, протекают с выделением тепла, но, поскольку коксование сопровождается и реакциями разложения, суммарный тепловой эффект отрицателен; в итоге пары, выходящие из камер,

Рис. 18. Установка с коксовыми камерами.
имеют температуру, на 30—50 °С ниже, чем температура ввода сырья в камеры.
Пары из камер, как упоминалось выше, проходят в колонну 6 (рис. 17), конструктивно оформленную по типу, описанному в гл. II (см. рис. 10, стр. 44): отгонная секция отсутствует, нижняя часть работает как конденсатор смешения и отделена от верхней части сборной тарелкой с горловиной. Предусмотрен отбор трех боковых погонов. Тарелки с S-образными элементами. В результате ректификации с верха колонны уходят пары бензина и воды н газ коксования, которые после конденсатора-холодильника 8 разделяются в водогазоотделителе 10 на водный конденсат, стекающий в сборник 13, на откачиваемый насосом 11 нестабильный бензин и жирный газ. Нестабильный бензин частично подается насосом И на орошение колонны 6, а балансовое количество, как и жирный газ, поступает во фракционирующий абсорбер 16, где происходит отделение сухого газа, т. е. частичная стабилизация бензина. Бензин с низа абсорбера 16 направляют на стабилизацию в колонну 18, с верха которой выводится отгон стабилизации (бутан-бутиленовая и частично пропан-пропиленовая фракции), а с низа — стабильный бензин коксования. Боковые погоны выводят из колонны 6
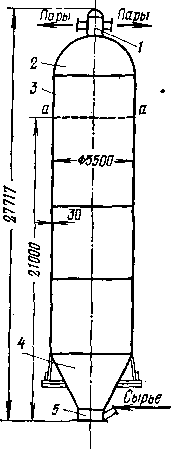
Рис. 19. Коксовая камера:
Рис. 20. Последовательность операций при
выгрузке кокса:
а — высверливание скважины; б — образование скважины; в — резка кокса.
Л
1,5 — верхняя и нижняя горловниы; 2 — полушаровое днище; 3 — цилиндрический корпус; 4 — коническое диище; а — а — линия максимального уровня кокса.
через секции отпарной колонны; с низа этих секций насосами 12 и 14 откачивают соответственно фракции дистиллята коксования — керосин, легкий и тяжелый газойли. Тепло отходящих потоков используют в теплообменниках 13 и 16 (нагрев нестабильного бензина 'и пара-турбулизатора) и рибойлере 15 фракционирующего абсорбера 20. Водяной конденсат из емкости 12 используют для производства водяного пара (в нижней части конвекционных труб печей 3 и 4).
Внешний вид подобной установки показан на рис. 18. Коксовые камеры представляют собой цилиндрические вертикальные аппараты (рис. 19), рассчитанные на давление от 0,18 до 0,6 МПа. Они имеют внутренний диаметр 4,6—5,5 м и высоту 27—28 м. Камера состоит из цилиндрического корпуса и двух днищ—сферического и нижнего конического, снабженных горловинами <и штуцерами.
Изготовлена камера из двухслойной стали; внутренняя легированная облицовка имеет толщину 2—4 мм. Камеры заполняют коксом попарно на 4/s высоты; продолжительность заполнения зависит от коксуемости исходного сырья и составляет от 24 до 36 ч. После заполнения коксом двух работающих камер их отключают от системы четырехходовыми кранами, позволяющими переключать поток сырья из печей без нарушения его непрерывности.

Рис. 21. Гидродолото (а) и гидрорезаки (б и в).
При переключении подачи сырья с одной камеры на другую необходимо предварительно прогреть включаемую камеру. Для этого ее вначале прогревают водяным паром, а затем 'направляют некоторое количество паров продуктов коксования из действующей камеры в верхнюю часть камеры, подготовленной к включению; пары проходят ее сверху вниз и в виде конденсата или паро-жидкостной смеси поступают в емкость, откуда жидкость откачивают в колонну. По мере прогрева камеры в емкости появляются пары, которые 'начинают поступать в колонну. При прогреве верхняя задвижка камеры полностью открыта для сообщения этой камеры с работающей, а долю паров, поступающих на прогрев, регулируют нижней задвижкой, сообщающей камеру с колонной. Когда работающая камера заполнилась коксом (камера, подготавливаемая к включению, должна к этому времени прогреться до 350— 360 °С), поток сырья переключают. При этом камеру, заполненную коксом, подготавливают к разгрузке: ее в течение 30—60 мин продувают паром, чтобы из коксовой массы удалить нефтяные пары. Эти пары направляют в колонну, а к концу продувки через емкость с газоотводящей трубой выводят в атмосферу. После охлаждения верхней части камеры до 200—250 °С в нее для охлаждения кокса подают воду до тех пор, пока не прекратится испарение воды в нагретой камере (об этом судят по появлению воды в сливной трубе емкости).
Для выгрузки кокса применяют гидравлический метод, заключающийся в использовании режущей силы водяных струй, подаваемых под давлением 10—15 МПа. Для этого над камерами устанав-лены буровые вышки высотой л; 40 м для укрепления бурового оборудования. После охлаждения камер открывают верхний и нижний люки и приступают к удалению кокса:
1) высверливают гидродолотом центральную скважину в толще кокса (рис. 20) и затем расширяют ее для свободного прохода гидрорезака;
2) удаляют основную массу кокса, перемещая по камере гидрорезак (рис. 21 и 22) — приспособление, снабженное соплами (как и гидродолото), направляющими сильные струи воды на стенки камеры, покрытые коксом. На рис. 21 представлены гидрорезаки, используемые последовательно, а «а рис. 22 дан гидрорезак ГРУ-2 конструкции БашНИИ НП, используемый на отечественных установках.
Продолжительность цикла работы двух камер (или четырех при двух параллельно работающих) зависит от вида сырья (тем короче, чем больше выход кокса) и составляет от 48 до 60 ч. В первом случае на подачу сырья в камеру затрачивают 24 ч (сернистое смолистое сырье), а во втором 36 ч (малосернистое сырье). Ниже приводится примерный график (в ч) работы камер при 48-часовом цикле:
Для удаления кокса и воды из-под днища камер предусмотрен железобетонный скат, облицованный листовой сталью и расположенный под углом «30°. На площадке воду дренируют, а кокс загружают в самосвалы или вагонетки и вывозят с установки. Воду после отстоя и фильтрования можно использовать повторно. По более совершенной системе предусмотрено комбинировать удаление кокса с дренажем воды, сортировкой кусков по размерам (>25 мм, 25—6 и 6—0 мм) и дроблением особо крупных кусков (>250 мм).
При системе транспортирования кокса на установке замедленного коксования (рис. 23) поток воды и кокса поступает через нижние люки камер 1 в дробильный агрегат 2, где дробятся наи более крупные куски, чтобы максимальный их размер не превы шал 250 мм. Из бункера-накопителя 3 кокс ссыпается на ленточ ные конвейеры 4, 5, 7 и 8, снабженные дренирующими устройст вами, и далее элеватором подается в грохот 6, где сортируется на целевую фракцию (>25 мм), среднюю фракцию (25—6 мм) и мелочь (6—0 мм). Вода, дренированная с конвейеров, отстаивается в фильтре-отстойнике 14. Со склада 9 кокс через питатель 10 разгружается конвейером 11 в вагон 12.
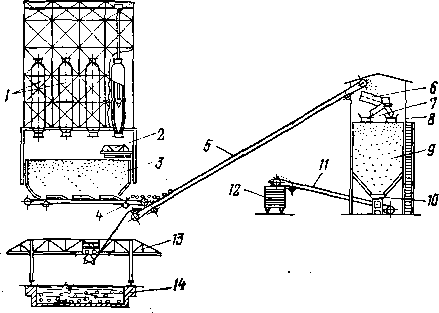
/ — крышка; 2 — узел переключения; 3 — корпус; 4 — успокоитель; S —режущее сопло: 6 — бурильные сопла.
Рис. 23. Система транспортирования кокса иа укрупненной установке замедленного коксования:
Cl
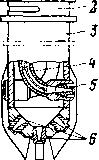
Рис. 22. Универсальный гидрорезак ГРУ-2;
1 — коксовые камеры; 2 — дробильный агрегат; 3 — буикер-иакопитель: 4, 5, 7, 8, 11— ленточные конвейеры; 6 — грохот; 9 — бункер; 10 — питатель; 12 — железнодорожный вагон; 13 — грейферный кран; 14 — фильтр-отстойник.
Установка замедленного коксования мощностью 1,5 млн. т сырья в год отличается от описанной следующими особенностями. Она имеет шесть коксовых камер и три трубчатых печи, каждая из которых обслуживает две камеры. Одновременно работают, таким образом, три камеры. Диаметр камер 7,0 м, высота 30 м. Температура нагрева сырья до 510°С, избыточное давление ~0,33 МПа. Вторичное сырье поступает в камеру из печей четырьмя потоками. Между камерами и колонной установлен эвапоратор-отбойник, чтобы предотвратить попадание в колонну мелких коксовых частиц, которые могут нарушить нормальную работу горячих насосов. На установке имеется еще одна печь — для циркулирующего газойля; он нагревается до 530°С и вносит тепло в камеру во время коксования и в первый период после отключения (с целью снижения содержания летучих в коксе). На установке используют конденсаторы-холодильники воздушного типа. Избыточное тепло направляют на производство водяного пара, а также в систему регенерации тепла в теплообменных аппаратах.
Основные показатели и факторы промышленного процесса!
Предусмотрена возможность работы камеры при двух давлениях} «0,4 МПа при переработке малосмолистого сырья и npi «0,18 МПа в случае высокосмолистого сырья. Малосмолистоё^ сырье дает меньше кокса и больше продуктов разложения, поэтск му во избежание чрезмерно большой скорости паров и переброса части содержимого камеры в колонну поддерживают давление более высоким. Так, при работе на малосернистой жирновской нефти выход кокса составляет всего 12,5%, а при работе на крекинг-остатке ромашкинской нефти он равен 33%; соответственно, суммарный выход газа, бензина и легкого газойля составляет 60,3% из малосмолистого сырья и 37,0% из смолистого.
При коксовании тяжелых га’зойлей термического или каталитического крекинга повышенное давление играет решающую роль, так как сырье при 495—510 °С на входе в камеру и при 440—460 °С на выходе и при давлении, близком к атмосферному, будет находиться в основном в газовой фазе, и выход кокса будет низкий. Например, при коксовании утяжеленного газойля термического происхождения в автоклаве при атмосферном давлении получено 7,9% кокса и 82,6% дистиллята; 9,5% приходилось на газ и потери; то же сырье, прококсованное при «0,3 МПа, дало 15,9% кокса, 70,2% дистиллята и 13,9% газа.
Большое влияние на выход и качество кокса оказывает коэффициент рециркуляции сырья. Обычно он составляет от 0,2 до 0,6. Более низкие значения соответствуют остаточному сырью (гудро-ны, остатки висбрекинга) при получении «рядового» кокса. При получении высокосортного «игольчатого» кокса используют ароматизированное дистиллятное сырье, >и разбавление его рецирку-лятом благоприятно влияет на качество кокса, так как рецирку-лят, побывавший в зоне коксования, содержит даже больше тяжелых ароматических углеводородов, чем исходное сырье.
Качество кокса заметно зависит от температуры нагрева сырья в печи. Чем выше температура на входе в камеры, тем меньше содержание летучих в коксе и тем выше его механическая прочность. Однако эта температура ограничена возможностью закоксо-вывания труб, а также образованием в камере некондиционного «гроздьевидного» кокса. Зависимость показателей качества кокса от температуры нагрева сырья в печи дана ниже:
| Прн 490 “С | При 500 “С |
При 510 °С |
| 10,0 |
7,8 | 6,8 |
| я 5 | «8 |
и 10 |
|
700 | 850 |
950 |
| 50 |
40 | 33 |
Показатели
Выход летучих, % .
Временное сопротивление раздавливанию, МПа Кажущаяся плотность, кг/м3 Пористость, %.....
гт^ков^стета^л^ет0^!2264°^ И ц-РЬЯ* В Камеру для пР™огонных остатков составляет и, и—0,13 ч , а для крекинг-остатков Гот
брекинга тяжелого сырья) 0,08—0 1 ч~1
Прокаливание кокса. Некоторые установки замедленного кок
сования снабжены лрокалочными агрегатами для удаления из кпк
са основной части так называемых летучих.
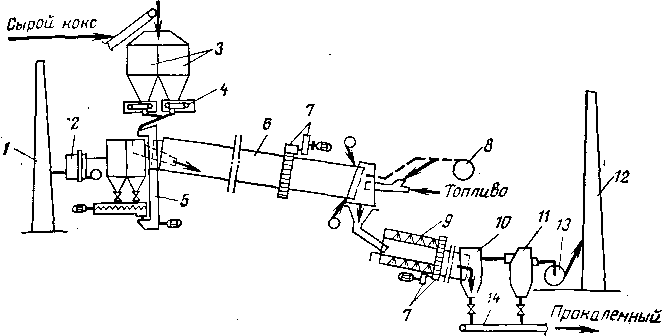
нонс
Рис 24 Схема прокаливания кокса во вращающейся горизонтальной печи:
' „„„„вяя труба: г —камера дожигания пыли; 3 — бункеры сырого кокса; ¦# —
/ ~ главная дымовая р^цевь]й элеватор. ^ _ прокалочная печь; 7 — система кольцевого питатели-дозеры, О 8 — воздуходувка; 9 — оросительный водяной холодильник; 10 —
воздушного осажден кокса- И — сборник коксовой пыли; 12 — вспомогательная труба для ^оКсТгазРоГизЛ сисГМы; 13 - вентилятор отсоса газа.
Выход летучих определяется количеством паров и газов, выделяющихся пои нагревании и выдерживании кокса при определен-ньтх^тяндаптных условиях (по ГОСТ 3929—65 прокаливание кокса ведут вТеТеме iмин при 850 "С). Максимально допустимое содержание летучих 7% (масс.) для кокса 1-го сорта и 9% (масс.) для кокса 2-го сорта. Так как кокс замедленного коксования откладывается в камерах при относительно низких температурах, то указанное содержание летучих не всегда удается выдержать. Частичного снижения летучих (на 1—1,5%) можно достигнуть, подавая после заполнения камеры коксом горячие (500 С и выше) папы газойля, смешанные с водяным ларом-тур-булизатором*. Однако для последующей подготовки кокса к использованию в ппоизводстве анодной массы или графитированных электродов его необходимо прокалить при 1200-1300 °С. В результате прокаливания повышается относительное содержание углерода в коксе, увеличивается его истинная плотность и снижается электрическое сопротивление.
Иногда прокаливание проводят на заводе-изготовителе электродной продукции, однако в'последние годы все более широко начинают применять прокалочные печи на нефтеперерабатывающих заводах'—при установках коксования. При этом можно «а 5—10% снизить количество кокса, транспортируемого с НПЗ, и облегчить его дальнейшее использование, так как при повышенном содержании летучих приходится сжигать их и а месте производства продукции, что усложняет технологию и повышает загазованность про-калочных цехов. Прокалочные агрегаты размещают на нефтеперерабатывающих заводах на открытом воздухе.
Наиболее распространены вращающиеся горизонтальные прокалочные печи (рис. 24). Печь 6 имеет небольшой наклон, позволяющий коксу ссыпаться к холодильнику 9 противотоком к продуктам, образующимся при сгорании топлива и к выделяющимся в печи летучим. Сырой кокс из бункеров 3 через дозирующие устройства 4 поступает в прокалочную печь, проходит ее и, охлаждаясь двумя вентиляторами 7, расположенными по окружности печи, и в оросительном холодильнике 9, выводится в бункер 10. Оттуда он ленточным конвейером 14 передается в емкость прокаленного кокса. Предусмотрено улавливание коксовой пыли (горячей, захваченной продуктами Сгорания и возвращаемой снова в печь, и холодной) после бункера 10 и сборника 11. Газы из этих емкостей отсасываются вентилятором 13 в дымовую трубу 12. Продукты сгорания после дожигания пыли в камере 2 уходят в главную дымовую трубу 1. Печи эти очень громоздки, например печь, дающая « 100 тыс. т прокаленного кокса в год, имеет длину « 60 м и наружный диаметр 4,3 м. Печь футерована огнеупорным материалом и имеет специальные внутренние устройства для регулированиЯ| движения кокса и времени прокаливания. I
Для осуществления полностью непрерывного процесса коксования! необходимо аппаратурное оформление, которое позволяло бы не-! прерывно выводить образующийся кокс из реактора. Идея непре-1 рывного вывода кокса (в виде небольших гранул) привела к раз-1 работке реактора с псевдоожиженным слоем кокса, выводимого в| виде порошка. В реакторном блоке в качестве теплоносителя цир-1 кулируют коксовые частицы, которые в результате контакта с! сырьем покрываются тонким слоем вновь образующегося кокса.I Некоторое количество частиц, наиболее укрупненных за счет мно-1 гократного «обрастания», непрерывно выводят из системы.
Коксование происходит на поверхности частиц кокса-теплоносителя. Вступая в контакт с горячей поверхностью частиц, сырье растекается по поверхности тонкой пленкой. Летучие продукты
коксования удаляются с поверхности и могут подвергаться последующему разложению, глубина которого зависит от длительности их пребывания в реакционной зоне.
Исследование кинетики непрерывного коксования в псевдоожи-женном слое порошкообразного кокса показало265, что этот процесс следует рассматривать как трехстадийный:
1) собственно коксование, сопровождающееся образованием основной массы продуктов разложения и уплотнения;
2) сушка или прокаливание кокса, в результате чего удаляются, летучие;
3) вторичные реакции распада и уплотнения продуктов коксования, находящихся в газовой фазе.
Скорость первой стадии весьма значительна и намного превышает скорость второй стадии. Так, для разложения гудрона ро-машкинской нефти при 500 °С на 80% необходимо всего 30 с, а для получения после этого кокса и доведения содержания в нем летучих до 0,2% требуется еще 2 мин. При этом интенсифицировать первую стадию процесса повышением температуры значительно легче, так как температурный коэффициент скорости этой стадии при 480—500 °С составляет 1,5—1,6, а для второй стадии всего 1,2—1,25. Исследование кинетики третьей стадии показало, что при 500 °С состав паров заметно изменился только через 30 с, а при 530 °С всего за 10—15 с.
Частицы коксового теплоносителя (рис. 25) имеют плотную слоистую структуру. Высокая температура кокса-теплоносителя и подача водяного пара в слой частиц способствуют испарению продуктов разложения и ускоряют их удаление с поверхности частиц, предотвращая тем самым образование продуктов вторичного уплотнения. Поэтому выход кокса при непрерывном коксовании меньше, чем при замедленном.
Ниже приведен материальный баланс (в % масс.) непрерывного коксования 28%-го гудрона ромашкинской нефти при 533 °С и длительности пребывания паров в реакторе 10 с:
Высокая кратность циркуляции теплоносителя сопряжена со значительными эксплуатационными расходами, но с другой стороны, повышенные температуры его нагрева достигаются сравнительно легко. Поэтому экономичнее придерживаться низкой кратности циркуляции. При коксовании минимальная кратность циркуляции должна быть такой, чтобы предотвратить слипание частиц теплоносителя при контактировании с сырьем. Большое значение при этбм имеет удельная поверхность частиц (т. е. поверхность на единицу массы). Так, при среднем диаметре частиц 0,25 мм поверхность 1 кг частиц (при плотности частиц 1000 кг/м3) равна «24 м2, а при среднем диаметре 7 мм всего 0,86 м2. Отсюда следует, что при наличии мелких частиц теплоносителя кратность его циркуляции

Рис. 25. Частицы порошкообразного кокса:
а — внешний внд; б — структура.
может быть умеренной. Для коксования в псевдоожиженном слое этот показатель не превышает 7—8 кг/кг.
Идея вести коксование в псевдоожиженном слое возникла в советской и зарубежной нефтеперерабатывающей промышленности позднее, чем аналогичный процесс каталитического крекинга. Первые промышленные установки были введены в эксплуатацию только в середине 50-х годов.
Технологическая схема установки и основная аппаратура. Принципиальная схема установки коксования в псевдоожиженном слое показана на рис. 26. Кокс-теплоноситель (порошок из округлых частиц диаметром 0,075—0,3 мм) непрерывно циркулирует через систему реактор 2 — коксонагреватель 3. Оба аппарата работают по принципу псевдоожиженного слоя. В реакторе псевдоожижение слоя кокса происходит путем подачи вниз водяного пара и частично при помощи паров и газов, образовавшихся при коксовании. В коксонагревателе псевдоожижение создают, подавая воздух; при этом часть кокса сгорает, а остальная масса нагревается до 600—620 °С.
На установке нет трубчатой печи для подогрева сырья — сырье поступает в реактор при умеренной температуре (300—350 °С) непосредственно с вакуумной установки «ли после нагревания в теплообменниках. Сырье подают в реакционную зону через систему распылителей под уровень псевдоожиженного слоя кокса. Пары и газы коксования проходят через систему циклонных сепараторов для отделения коксовой пыли и поступают в скруббер — парциальный конденсатор I, который для уменьшения закоксовывания передаточных линий расположен непосредственно на реакторе. За счет тепла циркуляционного орошения в нижней части скруббера конденсируется наиболее тяжелая часть дистиллята коксования;
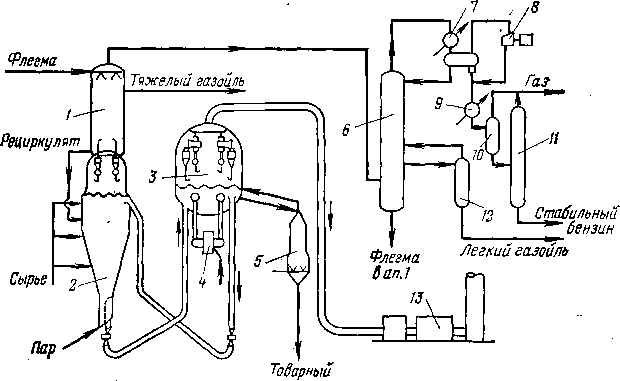
КОКС
Рис. 26. Схема установки непрерывного коксования в всевдоожижеином слое кокса:
/ — парциальный конденсатор (скруббер); 2 — реактор; 3 — коксонагреватель; 4 — топка; 5 — холодильник-классификатор кокса; 6 — ректификационная колонна; 7 — конденсатор; 8 — компрессор; 9 — холодильник; 10— газосепаратор; И — стабилизатор; 12— отварная колонна; 13 — котел-утилизатор.
ее возвращают в реактор 1. Дальнейшее погоноразделение (на газ +бензин и целевые газойлевые дистилляты) обычно происходит в отдельной ректификационной колонне 6.
Поток кокса непрерывно выводится из коксонагревателя через холодильник-классификатор 5. Классификация частиц по размерам (с выводом наиболее крупных в виде балансового кокса, с частичным дроблением и возвратом в систему мелких частиц, или «затравки») осуществляется внутри реакторного блока.
Реактор коксования представляет собой аппарат сложной конфигурации и переменного сечения (рис. 27). Псевдоожиженный слой кокса размещен <в конической 7 и цилиндрической 6 частях аппарата. Коническая форма нижней части аппарата способствует Уменьшению расхода пара на псевдоожижение. Внизу имеется отпарная секция 3; в нее подают снизу водяной пар для отпаривания порошкообразного кокса-теплоносителя от углеводородных продуктов коксования. В отпарной секции имеются полки для увеличения пути прохождения коксом отпарной секции. Форсунки для подачи сырья распылением его в слое кокса размещают обычно по высоте слоя в несколько ярусов. На крупных установках число форсунок достигает 100.
Верхняя часть 5 реактора имеет суженное сечение; уровень слоя может колебаться от низа до верха переходного конуса 2. Верх-
Пары
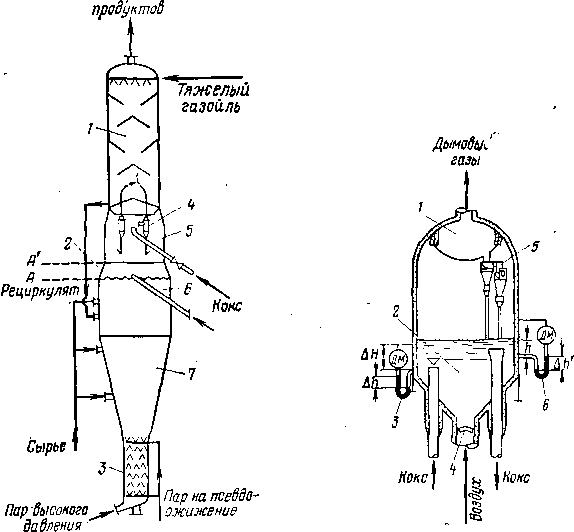
Рис. 27. Реактор коксования в псевдоожиженном слое:
А —А' — граница слоя; / — скруббер; 2 — переходный конус; 3 — отпарная секция; 4 — циклоны; 5 — зона газовой фазы; 6, 7 — зона псевдоожиженного слоя.
Рис. 28. Коксонагреватель:
/ — сборная камера; 2 — псевдоожиженный слой; 3, 6 — дифференциальные манометры; 4 — распределительная решетка; 5 — циклон.
няя часть заполнена парами продуктов коксования, содержащими некоторое количество коксовых частиц. Для удаления этих частиц служит система циклонов 6\ отделенные частицы возвращают по стоякам в слой. Сужение верхней части реактора обусловливает увеличение скорости паров над слоем; это позволяет, во-первых, свести к минимуму вторичное разложение и, во-вторых, повысить концентрацию частиц в парах: частицы, механически воздействуя на устья циклонов, предотвращают закоксовывание. Закоксовыва-
ние предотвращается также подачей горячей «струи» частиц кокса из коксонагревателя к устьям циклонов.
В нижнюю часть отпарной секции 3 предусмотрена подача пара высокого давления. При расширении пара, проходящего через сопло, укрупнившиеся частицы кокса дробятся; тем самым регулируется гранулометрический состав циркулирующего теплоносителя. Корпус реактора изготавливают из простой углеродистой стали и футеруют торкрет-бетоном. Футеровка толщиной «200 мм состоит из двух слоев — изоляционного слоя, который наносят на внутреннюю стенку корпуса и укрепляют затем сеткой, и из огнеупорного слоя толщиной «50 мм, заполняющего ячейки армирующей решетки.
Коксонагреватель имеет более простую конструкцию (рис. 28). Псевдоожижение коксовых частиц осуществляют воздухом, подаваемым через распределительную решетку 4. Как и в реакторе, корпус коксонагревателя футерован. Уровень горящего слоя кокса поддерживают постоянным с помощью сливного колодца, через край которого избыток кокса непрерывно уходит из аппарата. Для улавливания коксовой пыли в верхней части коксонагревателя имеются двухступенчатые циклоны 5; дымовые газы из циклонов поступают в котел-утилизатор.
Для разогрева системы при пуске служит топка под давлением, расположенная вблизи коксонагревателя; в ней сжигают жидкое или газообразное топливо. После предварительного разогрева системы при помощи топки зажигают форсунки, вмонтированные в стенки коксонагревателя, и продолжают 'разогрев до тех пор, пока не будет достигнута температура самовоспламенения кокса; после этого температура слоя в коксонагревателе начнет быстро возрастать, и форсунки станут не нужны.
Кокс циркулирует между реактором и коксонагревателем по U-образным линиям пневмотранспорта. Транспортирующим агентом служит водяной пар; транспортирование осуществляют без регулирующих задвижек, по принципу потока высокой концентрации. Коксопроводы имеют весьма значительный диаметр. Так, для установки с циркуляцией 3000 т кокса в час внутренний диаметр коксопровода равен «1 м. Чтобы предохранить коксопроводы от эрозии, их, подобно аппаратам реакторного блока, футеруют торкрет-бетоном.
На рис. 28 схематически изображены некоторые контрольные приборы, присущие системам с псевдоожиженным слоем.
Дифференциальный манометр 3 показывает перепад давления в слое. Если перепад равен Дh (м вод. ст.), а расстояние между точками отбора давления Равно ДН (м), то очевидно, что плотность слоя во столько раз меньше плотности воды, во сколько раз ДА меньше АН, и плотность слоя будет равна (в кг/м3):
Дифференциальный манометр 6 служит для определения высоты слоя. Если учесть, что перепад давления будет обусловлен только высотой слоя между замеряемыми точками, то высота слоя от нижней точки составит (принимая прежние обозначения и размерности для h и р и считая, что плотность слоя определена по манометру 3) :
Д h'
h = —т— 1000
Г
Большим достоинством коксования в псевдоожиженном слое является возможность вести процесс на установках высокой мощности. В настоящее время эксплуатируются установки с пропускной способностью по сырью ~7 тыс. т в сутки (более 2 млн. т в год), а с учетом рециркуляции — до 10 тыс. т в сутки. Установки выдают ежедневно до 1600 т порошкообразного кокса. При этом габариты аппаратов реакторного блока невелики для такой огромной мощности установки — внутренний диаметр реактора 8,9 м, а нагревателя 13,7 м; наивысшая точка установки расположена на 76 м от уровня земли. Избыточное тепло продуктов сгорания, уходящих из коксонагревателя, используют в мощном котле-утилизаторе, где образуется до 200 т пара высокого давления («4 МПа) в час. Установка расходует почти вдвое меньше пара, чем произ-
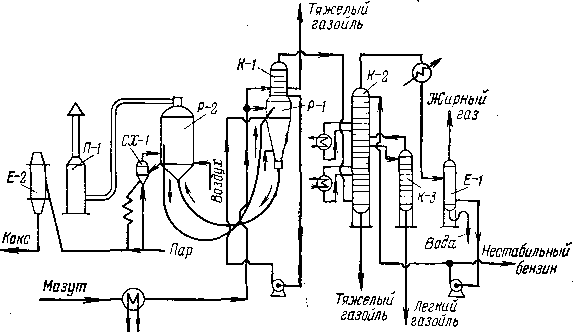
Рис. 29. Схема блока ТКК комбинированной установки АТ-ТКК:
р-1 _ реактор; Р-2 — коксонагреватель; СХ-1 — отвеиватель; П-1 — котел-утилизатор; К-1 — парциальный конденсатор; К-2 — ректификационная колонна; К-3 — отпариая колонна; Е-1 — водоотделнтель-газосепаратор; Е-2 — емкость кокса.
водит, т. е. помимо основного назначения она выполняет роль дополнительной котельной.
Значительные ресурсы избыточного тепла 'на установке коксования позволяют успешно комбинировать этот процесс с прямой перегонкой и другими процессами переработки нефти, требующими затрат тепла. На рис. 29 приведена технологическая схема блока коксования комбинированной установки атмосферной перегонки и коксования (АТ-ТКК) мощностью 3 млн. т нефти в год.* Отличительная особенность установки — использование избыточного тепла, получаемого в блоке коксования, для прямой перегонки мазута.
Блок атмосферной перегонки имеет обычную схему двухступенчатого подогрева нефти: нагретая в теплообменных аппаратах нефть проходит колонну предварительного испарения, где отделяются бензиновые фракции. Полу мазут подогревают в печи и подают в основную ректификационную колонну, с низа которой отводят горячий мазут.
При поступлении горячего мазута с низа основной колонны в парциальный конденсатор К-1 (рис. 29) от мазута отгоняется широкая фракция; одновременно конденсируется и охлаждается часть парообразных продуктов коксования, выходящих из реактора Р-1 при 510—540 °С. Остаток в нижней части парциального конденсатора К-1 представляет собой, таким образом, смесь гудрона и тяжелого рециркулята, которая является загрузкой реактора Р-1. С низа колонны К-2 (где разделяются продукты коксования) уходит фракция, состоящая из тяжелого отгона (от мазута) и тяжелого газойля коксования. Легкий газойль отбирают в виде бокового погона из колонны К-2 (через колонну К-3) \ газ и пары бензина уходят с верха колонны К-2 и после охлаждения в конденсаторе-холодильнике разделяются в газосепараторе Е-1.
Схема позволяет отказаться от сооружения вакуумной трубчатой установки в том случае, когда нефть перерабатывают по топливной схеме, а широкую тяжелую фракцию направляют на каталитический крекинг.
Ниже приводится типичный технологический режим блока кок
сования в псевдоожиженном слое кокса:
Реактор
Температура в слое, °С........’ 510—540
Давление над слоем, МПа..............0,14—0,16
Массовая скорость загрузки, кг/(кг-ч) .... 0,6—1,0
Длительность пребывания кокса-теплоносителя, мии
Скорость паров над слоем, м/с............0,3—0,5
Кратность циркуляции кокса, кг/кг..........6,5—8,0
Коксонагреватель
Температура в слое, °С.......600—620
Давление над слоем, МПа..............0,12—0,16
Длительность пребывания кокса-теплоносителя
в слое, мин......................6—10
Скорость дымовых газов над слоем, м/с .... 0,5—0,7
Интенсивность горения кокса, кг/ч на 1 т слоя . . 30—40
^ Голов Г. С. — Новости нефтяной техники. Сер. Нефтепереработка, 1961,
При анализе простой и комбинированной схем (рис. 26 и 29) видно, что тепло, расходуемое на нагревание сырья до температуры коксования и на реакцию крекинга, получается в результате сжигания в коксонагревателе Р-2 большей или меньшей доли образующегося (балансового) кокса.
Материальный и тепловой ба-Пводипты. , _ Коксовый лансы блока коксования показы
шсобания
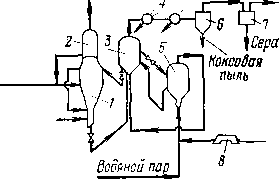
Воздух
Сырье
Водяной
пар
Рис. 30. Схема процесса флексико-кинг:
/ — реактор коксования; 2 — парциальный конденсатор; 3 — коксонагреватель; 4 — котел-утилизатор; 5 — реактор газификации; 6 — циклон; 7 — секция сероочистки; 8 — воздуходувка.
вают, что при переработке гудрона с плотностью «1000 кг/м3 количество кокса, сжигаемого в коксонагревателе, примерно в четыре раза меньше, чем суммарный выход кокса. Таким образом, потенциально установка коксования может работать без вывода кокса — с полным сжиганием всего балансового его количества внутри системы.
Использование порошкообразного кокса. Сернистый порошкообразный кокс употребляют в основном как топливо и не используют для электротермических процессов. На некоторых зарубежных заводах этот кота размалывают до пылевидного состояния и сжигают в тбпках печей для прямой перегонки нефти, в котельных и др. Учитывая высокое содержание серы в порошкообразном коксе, вызывающее образование значительных количеств SO2 в продуктах сгорания, было предложено этот кокс газифицировать266 (рис. 30).
Для этой цели кокс из, коксонагревателя направляют в реактор газификации, где при высокой температуре кокс обрабатывают воздухом (кислородом) и водяным паром и превращают в так называемый коксовый газ, состоящий из смеси водорода, оксида и диоксида углерода, сероводорода и паров воды. Коксовый газ очищают от H2S и используют как топливо, хотя его теплота сгорания невелика — всего 3800—4800 кДж/м3.
Гибкость коксования в псевдоожиженном слое позволяет использовать процесс в самых разных вариантах. Возможность подогрева сырья без трубчатых печей (с легко закоксовывающимися трубами) позволяет использовать как сырье коксования даже остатки типа битумов с плотностью выше 1000 кг/м3. Коксование в
¦ псевдоожиженном слое используют в основном применительно к
высокосернистому остаточному сьгрью. В этом случае в коксе концентрируются тяжелые металлы и до 30—40% серы, содержащейся в сырье. Образующиеся дистилляты можно подвергать каталитической переработке (например, гидрокрекингу) с меньшими эксплуатационными затратами, чем при непосредственной переработке нефтяных остатков.
Материальный баланс и качество продуктов. Выход кокса в процессе коксования определяется в основном коксуемостью сырья и практически линейно изменяется в зависимости от этого показателя.
Максимально возможный выход кокса из данного сырья достигается в том случае, когда все образующиеся газойлевые фракции возвращают (в качестве рециркулята) на повторное коксование. Такая форма коксования называется крекингом до кокса; конечные продукты — газ, бензин и кокс. Промышленного применения этот процесс не имеет, так как широкую газойлевую фракцию экономичнее -использовать для каталитического или гидрокрекинга с получением продуктов более высокого качества.
При замедленном коксовании высокосмолистых остатков выход кокса составляет 1,5—1,6 от коксуемости сырья (определенной стандартным методом):
и>к= 1,5 ч-1,6К-
Выход остальных продуктов коксования определить сложнее, поскольку начало кипения рециркулята — величина переменная, а в связи с этим изменяется и конец кипения самого тяжелого из отбираемых дистиллятов. 3. И. Сюняев предлагает 'использовать для определения выхода бензина эмпирическое уравнение:
-V
Рост — 0,940
0,00019 ( '
где Б — выход бензина (40—205 °С), % (масс.) на сырье; рост — относительная плотность исходного остаточного сырья.
Уравнение выведено применительно к сырью с плотностью от 0,960. До 1,000 и при условном начале кипения рециркулята 500°С.
По В. JI. Нельсону, выходы бензина и керосино-газойлевой .фракции связаны уравнением:
248
-Б
Ъ = ~?т--<22>
^Г-169
где В — выход керосино-газойлевой фракции (до 510вС), % (об.); рг и р0Ст — соответственно плотность керосино-газойлевой фракции и сырья; Б — выход бен. зииа (до 204°С), % (об.).
Если определить выходы кокса, бензина и широкой керосино-газойлевой фракции, можно по разности установить выход газа (плотность сырья известна, а плотность бензина и газойля можно взять из опытных данных):
Г = 100 — swK — б — В
Подобный подсчет, однако, будет приближенным.
Выше отмечалось, что наилучший кокс (игольчатой структуры) получается из дистиллятного сырья. Выход этого кокса весьма значителен благодаря высокой концентрации в нем полициклических ароматических углеводородов и совершенно не соответствует коксуемости сырья, так как при этом стандартном испытании более легкое сырье частично испаряется из тигля, и выход кокса в промышленных условиях несопоставим с коксуемостью. Например, фенольный экстракт газойля коксования, содержавший «66% ароматических углеводородов и 16,7% смол, имел коксуемость 3,6%, а выход кокса из него на проточной лабораторной установке (при «0,1 МПа) составил 19,1%.
Материальные балансы замедленного и непрерывного коксования весьма различаются. В табл. 7 приведен выход кокса при различных способах коксования, включая и крекинг до кокса. При непрерывном коксовании выход кокса приближается к коксуемости сырья, составляя в среднем 110—115% от этой величины. При замедленном коксовании выход кокса значительно превышает коксуемость и достигает 150—180% от нее. В табл. 8 представлены усредненные материальные балансы отечественных установок замедленного коксования при работе на различном сырье.
Разница в выходе кокса обусловлена тем, что при непрерывном коксовании вследствие высокой температуры теплоносителя образующиеся продукты разложения быстро удаляются с его поверхности. Часть тяжелых продуктов первичного распада не успевает ни Таблица 7. Выход кокса нз различного сырья
Данные В. J1. Нельсона
| Коксуемость сырья, % (масс.) | Относительная плотность | Выход кокса, % (масс.) | |||
| сырье промежуточного основания |
сырье асфальтеио-вого основания и крекинг-остаток | замедленное коксование | коксование в псевдоожиженном слое |
крекинг дикокса | |
|
5 | 0,901 |
0,925 | 8,5 |
3,0 | 22 |
|
10 | 0,962 |
0,975 | 18,0 |
11,5 | 37 |
|
15 | 1,003 |
1,017 | 27,5 |
17,0 | 40 |
|
20 | 1,031 |
1,043 | 35,5 |
23,0 | 45 |
|
25 | 1,052 |
1,062 | 42,0 |
29,0 | 48 |
|
30 | 1,062 |
1,079 | — |
34,5 | — |
|
40 | - | 1,100 | — | 46,0 |
— |
Таблица 8. Материальный ба^нс установок замедленного коксования при работе на разливом сырье
Данные 3. И. Сюняева
|
сг Л | О | Выход, % (масс.) | ||||||
| Сырье | 3 V А Н V О ? §<= с^* | Коксуемость сырья, % (мае | Я « U | бензин | (н. к.—205 °С) |
легкий газойль (205—350 °С) | 1 а с-. * о %%Л Е- СО —» | и К О ? | Потери*, % (масс.) |
| Мазут | 0,950 |
9,9 | 4,5 | , 7,5 |
40,0 | 29,0 | 15,0 | 4,0 |
|
Полугудрон | 0,965 |
12,6 | 6,0 | 12,0 |
30,0 | 26,0 | 22,0 | 4,0 |
| Г удрои Крекинг-остаток | 0,991 |
16,0 | 7,0 | J6,0 | 26,0 | 23,0 |
24,0 | 4,0 |
|
облегченный | 1,012 |
20,0 | 7,5 | 7,0 |
17,0 | 32,0 | 32,0 | 4,5 |
|
утяжеленный | 1,024 |
23,0 | 8,0 | t-,8 |
12,0 | 35,0 | 35,0 | 3,2 |
* Завышенные потери объясняются сбросом части дистиллята в атмосферу при пропаривании кокса и потерями дистиллята во время прогрева коксовых камер.
разложиться на более легкие фракции, ни образовать дополнительные смолисто-коксовые отложения на частицах теплоносителя и удаляется из зоны реакции 'в виде паров вместе с другими продуктами разложения. Поэтому по фракционному составу жидкие продукты непрерывного коксования, как правило, тяжелее, чем про-
Таблнца 9. Качество жидких продуктов замедленного коксования
Данные А. И, Стехуна и соавт.
|
Бензин | Легкий га |
Тяжелый га | ||
|
Показателя | нз маигыш-лакской яефти | из западносибирской иефтн | зойль из западносибирской нефти | зойль из западносибирской иефти |
| Плотность р|° | 0,720 | 0,736 | 0,867 |
0,918 |
| Пределы выкипания, °С |
37—180 | 46—180 |
190—360 | 220—430 |
|
Бромное число, г Вг2/100 г | 72,7 | 77,2 |
¦— | — |
|
Содержание серы, % (масс.) |
0,09 | 0,54 | 1,70 |
2,10 |
| Октановое число (м. м.) | 58 |
63 | — | — |
| Цетановое число |
— | — | 45 |
— |
| Температура застывания, °С Групповой состав, % (об.) | — | — |
—22 | + 10 |
|
парафнно-нафтеновые в том числе | 45,9 | 36,6 |
— | — |
|
н-парафины | — |
— | 8,5 | 12,0 |
| изопарафины |
— | — | 19,1 |
7,9 |
| нафтены и циклоолефины | — |
— | 29,9 | 25,8 |
| ароматические |
5,5 | 12,7 | 34,4 |
39,2 |
| сернистые (тиофены) | — | — | 2,0 | 3,1 |
| олефины | 48,6 |
50,7 | — | — |
| смолы |
' | 6,1 | 6,8 |
|
дукты полупериодического и периодичккого коксования. Уточненный выход жидких продуктов и газа гри непрерывном коксовании Можно определить только опытным путем.
Качество продуктов коксования Д1Н0 в табл. 9. Видно, что бензин, получаемый при замедленном коксовании, имеет невысокое октановое число, повышенное содержание непредельных и для сернистого сырья — серы. Наиболее рщиональный путь его использования— глубокая гидроочистка с последующим каталитическим риформингом. Легкий газойль тагже нуждается в гидроочистке — как для снижения содержания ;еры, так и для удаления непредельных, после чего газойль м^жно использовать в качестве дизельного топлива. Тяжелый пзойль может явиться сырьем или компонентом сырья для производства «игольчатого» кокса (при ограниченном содержании сега) или идти на каталитическую переработку (каталитический крекинг, гидрокрекинг).
ПИРОЛИЗ НЕФТЯНОГО СЫРЬЯ
Пиролиз — наиболее жесткая форма термического крекинга нефтяного и газового сырья, осуществляемая обычно при 700—900 °С с целью получения углеводородного газа с высоким содержанием непредельных. Режим может быть направлен на получение максимального выхода этилена, пропилена или бутиленов и бутадиена. Наряду с газом образуется некоторое количество жидкого продукта — смолы, содержащей значительные количества’ моноцикли-ческих (бензол, толуол, ксилолы) и полициклических ароматических углеводородов (нафталин, антрацен). Долгое время, пока не был разработан процесс каталитического риформинга, пиролиз являлся практически единственным промышленным методом получения ароматических углеводородов из нефти.
В настоящее время целевым продуктом пиролиза является газ, богатый непредельными, из которых основная роль принадлежит этилену. О масштабах потребности химической промышленности в этилене и о росте спроса на него можно судить по следующим цифрам: в 1976 г. мировое производство этилена составляло 35 млн. т и предполагается, что в 1980 г. оно достигнет 52 млн. т267. Области использования этилена весьма разнообразны. Более 40% этилена расходуется на производство полиэтилена; значительную долю занимают производства этилового спирта и стирола (сырье для производства синтетического каучука).
Все большее значение приобретают и другие газообразные непредельные углеводороды. Например, в 1973—1977 гг. в странах Европейского Экономического Сообщества соотношение между объемами производства этилена и пропилена составляло «1,8: 1. Пропилен используют для получения полипропилена, высокооктанового компонента бензина (алкилированием изобутана), жидких полимеров С6—СJ2, используемых в качестве топливных компонентов (полимербензин), моющих средств и др. Бутадиен (дивинил), используемый в производстве синтетического каучука, получают не только путем пиролиза жидких нефтепродуктов, но и каталитическим дегидрированием н-бутана. Однако в последние годы доля бутадиена, вырабатываемого пиролизом, возросла, так как экономически этот процесс более благоприятен.
Выход смолы пиролиза тем больше, чем тяжелее сырье.
Сырье для пиролиза весьма различно. Пиролизу подвергают газообразные углеводороды (этан, пропад, бутан и их смеси) и жидкие (низкооктановые бензины, керосино-газойлевые фракции, нефтяные остатки). Выбор сырья определяется в первую очередь целью пиролиза. Для производства этиленсодержащего газа пригодно любое сырье, но наибольший выход этилена дает этан. Для получения высокой концентрации пропилена в газе пиролиз этана непригоден, так как этан в основном дегидрируется до этилена. Соответственно, для получения высоких выходов бутадиена не используют ни этан, ни пропан.
При выборе сырья учитывают также его ресурсы, связанные с направлением и уровнем развития нефтеперерабатывающей и газовой промышленности данной страны. Известно, что в США потребление бензина наиболее значительно из-за большого парка легковых автомашин, поэтому преобладающим сырьем пиролиза в этой стране до 70-х годов являлись природные газы. В это же время в Советском Союзе и странах Западной Европы глубина переработки нефти была значительно ниже. Повышение требований к моторным качествам бензинов дало возможность использовать как сырье пиролиза и газовый бензин и низкооктановые бензины прямой гонки. Одновременно в качестве сырья использовали и газообразные углеводороды природных и заводских газов. Пиролиз газообразного сырья (особенно этана) дает более высокие выходы этилена, чем пиролиз жидкого сырья.
С середины 70-х годов в связи с повсеместным углублением переработки нефти и ростом цен на бензин возникает интерес к пиролизу более тяжелого сырья — керосино-газойлевых фракций, вакуумного газойля. Был также разработан пиролиз сырой нефти с использованием перегретого до 2000 °С водяного пара в качестве теплоносителя.
Наиболее распространенная форма промышленного процесса — пиролиз в трубчатых печах. Совершенствование конструкций печей позволило расширить фракционный состав сырья, и к концу 70-х годов мощность единичных трубчатых установок промышленного пиролиза достигла 700 тыс. т этилена в год. Одновременна
Таблица 10. Типичный выход продуктов пиролиза различного сырья
Данные С. В. Зденека и др.
|
Пиролиз газового сырья |
Пиролиз жидкого сырья | |||||
|
Показатели | этан |
пропан | н-бутан |
бензин | газойль |
вакуумные газойли |
| Чистота сырья, % |
95,2 | 98,2 | 97,0 | _ | _ |
- |
| Плотность pi0 | — | 0,713 | 0,832 | 0,870 | ||
| Пределы выкипания, °С |
— | — | — |
32—171 | 232—327 |
300—538 |
| Характеризующий фактор | — | — |
— | 12,3 | 12,0 | 12,2 |
| Содержание ароматических уг | — |
— | -: | 7 |
24 | 28 |
|
леводородов в сырье, % | ||||||
|
|(масс.) | ||||||
| Выход, % (масс.) | ||||||
| этилен | 48,0 |
36,7 | 31,6 | 31,3 | 26,0 | 23,0 |
| пропилен | 2,1 |
14,0 | 17,8 | 12,1 |
9,0 | 13,7 |
|
бутилены | 1,1 | 3,1 | 2,4 | 2,8 |
2,0 | 4,9 |
|
бутадиен | — | — | 1,7 | 4,2 |
4,2 | 6,3 |
|
сухой газ | 8,4 |
33,2 | 32,2 | 18,2* |
16,2* | 11,2* |
|
ароматические Се—Cg | ) |
I | j | 13,0 |
12,6 | } 16,9 |
| бензин С5 — 204 °С (без | 1,7 | 1 5,0 | 1 103 | 9,0 |
8,0 | |
| ароматических) | 6,0 | 19,0 | 21,0 | |||
|
тяжелая смола (>204°С) | ) | J | J 4,0 | |||
| непревращенное сырье | 38,7 | 8,0 |
— | — | - | |
| Степень превращения сырья за | 60 | 92 | 96 | Без рециркуляции | ||
| один пропуск, % (масс.) |
||||||
* Плюс пропан.
возвращаются к не получившим вначале широкого распространения термоконтактным процессам пиролиза остаточного сырья и к пиролизу сырой нефти.
В табл. 10 и И представлен материальный баланс пиролиза различного сырья. Следует иметь в виду, что наряду с максималь-
Таблица И. Выход продуктов пиролиза и пропускная способность' по сырью для получения 300 тыс. т этилена в год
| Показатели |
Пиролиз эгана |
Пиролиз пропана |
Пиролиз бензина |
Пиролиз газойля |
|
Выход продуктов, % (масс.) на сырье |
33,7 | |||
| этилен | 77,7 |
42,1 | 25,6 | |
|
водород | 5,4 | 1,5 | 1,1 | 1,1 |
| сухой газ (СН4+С2Н6) | 8,5 | 25,8 |
17,2 | 12,2 |
|
фракция Сз | — |
22,1 | 15,5 | 10,5 |
| фракция С4 |
—: | — | 8,7 | 5,5 |
| легкое масло (до 200°С) | — |
—. | 18,2 | 24,6 |
| тяжелое масло |
— | — | 5,6 |
20,5 |
| Пропускная способность по сырью, тыс. т в год |
390 | 710 | 890 | 1170 |
ными выходами этилена использование газообразного сырья позволяет работать с рециркуляцией, т. е. с возвратом непревращенного этана или пропана (после установки газоразделения) в зону реакции. Для жидкого сырья это неприменимо, так как смола пиролиза является высокоароматизированным продуктом глубокого превращения, частично совпадающим по фракционному составу с сырьем, и возврат ее на повторный пиролиз вызвал бы закоксовы-вание труб печи.
Теоретические основы процесса
Пиролизу присуще глубокое преобразование исходного сырья, приводящее к легким газообразным углеводородам, к ароматическим моно- и полициклическим углеводородам, & также к продуктам глубокого уплотнения (кокс и сажа268).
Если обратиться к графику изменения энергии Гиббса для некоторых углеводородов в зависимости от температуры (рис. 31), станет очевидным, что в области высоких температур наиболее стабильны олефины и ароматические углеводороды. Выше 790°С этилен становится ста-бильнее этана (прямые для этана и этилена пересекаются); при еще бо-' лее высоких температурах (>1120°С) наиболее стабилен ацетилен; таким образом, интервал 790—
-62,16
300 500
700 soo поо
Температура, К
1300 1500
Рис. 31. Изменение энергии Гиббса для углеводородов в температурной области пиролиза.
1120°С является термодинамически возможным для получения этилена из этана. Аналогичные соображения для пиролиза пропана с целью получения этилена позволяют установить примерный температурный интервал от 660 до 930 °С.
Реакции образования ароматических углеводородов при пиролизе весьма многообразны. В результате термической сополимеризации непредельных образуются циклоолефины, которые далее дегидрируются до ароматиче-
ских углеводородов. Так 'протекает, например, взаимодействие бутадиена и этилена с образованием бензола:
Л
НС сн,
I + II -1
НС сн,
^сн,
Эта и подобные ей реакции, относящиеся к так называемому диеновому синтезу, свойственны пиролизу и обусловливают ароматизированный состав смолы.
Результаты пиролиза оценивают по выходу целевого продукта, например этилена. Поскольку основными факторами пиролиза являются температура и длительность реакции, каждой температуре соответствует некоторое оптимальное время контакта — при котором выход целевого продукта максимален (рис. 32).
Сравнительные данные по пиролизу различного газообразного сырья показывают, что выход ароматических углеводородов наиболее высок при пиролизе олефинов; это подтверждает их роль в образовании ароматических углеводородов. Аналогичное явление можно наблюдать и при пиролизе бензинов, содержащих непредельные углеводороды.
В табл. 12 представлены состав легкой части смолы при 'пиролизе бензина, получаемого замедленным коксованием, и (для сопоставления) концентрация ароматических углеводородов в смоле при пиролизе прямогонного бензина; данные получены при близких температурах и времени контакта (»1 с). Бензин замедленного коксования имел следующий групповой состав: 61,6% (масс.) парафино-нафтеновых углеводородов, 15,4% (масс.) ароматических и 23% (масс.) непредельных. Прямогонный бензин, естественно, не содержал непредельных и в основном .состоял из парафинонафтеновых. Как видно из таблицы, наличие непредельных углеводородов в бензине 'позволило увеличить выход ароматических углеводородов примерно на 7% (масс.) на исходное сырье. Однако это достигается за счет снижения газообразования и, следовательно, за счет уменьшения выхода целевых олефинов, что можно видеть
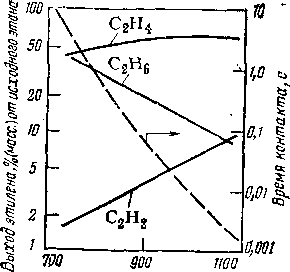
доо
Температурите
Рис. 32. Максимальный выход этилена при пиролизе этаиа в интервале 730—1100 °С.
для времени кон-
Пунктир
такта.
- кривая
Таблица 12. Групповой состав и содержание ароматических углеводородов во фракции н. к. — 150 °С смолы пиролиза
| Пиролиз бензина замедленного коксования* | Пиролиз прямогоиного беизииа** пси 780 °С | |||||
| Показатели | при |
750 °С | при 800 °С | |||
| иа смолу | на исходное сырье | на смолу | иа исходное сырье | на смолу |
иа исход-иое сырье | |
|
Выход смолы, % (масс.) |
— | 34,3 | _ |
37,6 | — |
22,0 |
| Содержание, % (масс.) бензол | 20,1 | 6,9 | 21,3 |
7,9 | 23,2 |
5,1 |
| толуол | 15,8 | 5,4 |
17,9 | 6,7 |
13,2 | 2,9 |
| ксилолы и этилбензол |
6,0 | 2,1 | 8,1 | 3,1 | 5,0 |
1,1 |
|
непредельные | 6,9 | 2,4 | 0,8 |
0,3 | Нет данных | |
| пар афино-нафтеновые | 3,0 | 1,0 |
0,1 | » ' |
||
* Данные Н. Д. Александровой н Е. В. Смидович. ** Данные Т. Н. Мухиной и сотр.
по выходу 'смолы, который значительно больше при пиролизе бензина, получаемого замедленным коксованием.
Наличие ароматических углеводородов в сырье пиролиза снижает газообразование из-за высокой термической стабильности этих углеводородов, однако их состав может изменяться за счет термического деалкилирования, при углублении процесса.
В связи с переходом на пиролиз утяжеленного сырья представляет интерес установить, как би- и полициклические ароматические углеводороды, присущие керосино-газойлевым фракциям, в сырье влияют на процесс. Установлено, что эти углеводороды тормозят образование легких олефинов, причем тормозящее действие бици-клических углеводородов слабее, чем для полициклических. При частичном удалении полициклических углеводородов повышается выход этилена и суммы газообразных олефинов. Тормозящее действие полициклических углеводородов может быть объяснено тем, что прочные би- и полифенильные радикалы присоединяют легкие свободные радикалы, образующиеся при распаде алифатических углеводородов, и обрывают цепи целевых реакций. В итоге выход этилена из утяжеленного дистиллятного сырья снижается, очевидно, не только за счет свойственных этому сырью реакций.
Энергия активации при процессах уплотнения ниже, чем при реакциях расщепления, поэтому пиролиз на олефины предпочтительно вести при высокой температуре и малом времени контакта. При завышении времени контакта образовавшиеся непредельные углеводороды тоже подвергаются реакциям уплотнения, что снижает выход целевых олефинов.
В соответствии с содержанием водорода в молекуле, наибольший выход газа (при минимальном коксообразовании) может дать газообразное сырье — этан, пропан, н-бутан. Из жидкого сырья наибольший выход газа дает бензин парафинового основания, с пониженным концом кипения, о чем свидетельствуют данные табл. 10 (стр. 110).
Для каждого вида сырья существует оптимальное сочетание температуры и продолжительности пиролиза. Например, из рис. 32 видно, что максимальный выход этилена из этана соответствует температуре «1000°С и времени контакта 0,01 с; при 900 °С максимум выхода этилена соответствует времени контакта 0,08 с. Однако выбор температуры на промышленной установке определяется не только изложенными соображениями, но и аппаратурным оформлением. Термоконтактный пиролиз можно осуществлять при более высоких температурах, а в трубчатых печах применение высоких температур (1000°С и более) затрудняется из-за необходимости подбора жароупорных материалов, забивания труб сажей и
¦ коксом, а также сохранением весьма малого времени контакта. Кроме того, имеет значение концентрация нежелательных компонентов в получаемом газе. Так, пиролиз углеводородного газа с Получением этиленз при 1000 °С сопровождается заметным образованием ацетилена; избежать этого можно при более мягком режиме.
«Этиленовому» режиму пиролиза соответствует пониженный выход пропилена, так как он при этом частично превращается в этилен. Наиболее мягкие «бутиленовый» и «бутилен-бутадиено-вый» режимы из-за наименьшей термической стабильности этих углеводородов. На рис. 33 показана кинетика коксообразования при пиролизе различного газообразного сырья в одинаковых температурных условиях (815°С). Для проведения пиролиза жидкого сырья с целью получения этилена требуется более мягкий температурный режим, чем при пиролизе газов.
Для сравнительной характеристики режимов пиролиза жидкого сырья и термической стабильности этого сырья по сравнению с другим часто используют фактор жесткости f, предложенный Лин-деном
f = /т°,об (23)
где t — температура (°С), at — время контакта (с). На рис. 34 показано изменение химического состава газов пиролиза в зависимости от фактора жесткости для нескольких -нефтяных и газоконденсатных фракций, элементный состав которых отвечает соотношению С : Н от 5,2 : 1 до 7,25 : 1. График свидетельствует о том, что эти фракции были близки по составу, так как точки для разны^ фракций ложились на общие кривые, и что выход этилена макси! мален при факторе жесткости от 800 до 830. I
Иногда фактор жесткости заменяют другими показателями —I используют атомное отношение Н : С в жидких продуктах пиролиза (Св и выше). Чем более жестким принят режим, тем, естественно, ниже это соотношение. Оно зависит и от качества сырья, а для данного сырья позволяет также сопоставлять эффективность конструкций трубчатых печей. Например, при пиролизе бензина в обычных печах на жестком режиме имеем Н:С = 1 : 1, а в печах
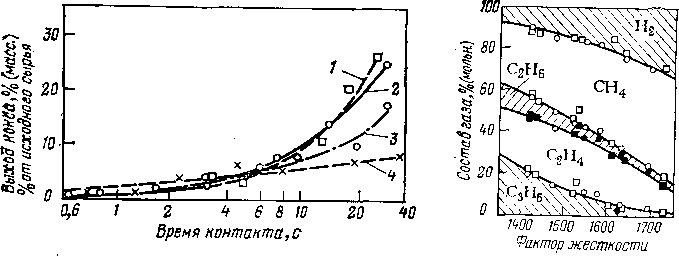
Рис. 33. Кинетика коксообразования при пиролизе различного газообраздого сырья!:
/ — этилен; 2 — пропилеи; J •— пропан; 4 — этаи.
Рис. 34. Изменение химического состава газов, образующихся при пиролизе га-зоконденсатных и нефтяных фракций при «0,1 МПа, в зависимости от фактора жесткости.
типа «миллисеконд» Н:С = 1,16:1, что соответствует выходу этилена (с рециркуляцией этана) до 35,3% (масс.).
При пиролизе газойлей хорошим критерием для определения максимального выхода этилена при жестком режиме служит индекс корреляции, характеризующий степень ароматичности сырья. Между индексом корреляции и максимальным выходом этилена существует линейная зависимость.
Значительное газообразование при пиролизе жидкого сырья и ¦образование более легких, чем сырье, компонентов при пиролизе газообразного сырья вызывают большое увеличение (обычно в 1,5—2 раза) удельного объема паровой (газовой) фазы. Известно, что реакциям, протекающим с увеличением объема, способствует низкое давление в реакционной зоне или, что то же самое, низкое парциальное давление продуктов. Для уменьшения роли реакций Уплотнения пиролиз ведут при максимально низком давлении. В случае применения трубчатых реакционных аппаратов на входе в змеевик приходится поддерживать некоторое избыточное давление, чтобы преодолеть гидравлическое сопротивление в трубах печи и в последующей аппаратуре (обычно на выходе из печи Давление равно 0,2—0,25 МПа).
Для компенсации отрицательного влияния этого давления и общего улучшения показателей процесса практикуется подача в печь вместе с сырьем перегретого водяного пара, снижающего парциальное давление паров продуктов пиролиза и препятствующего реакциям уплотнения. На рис. 35 представлены кривые выхода
этилена и кокса при пиролизе пропана в зависимости от разбавления пропана водяным паром. Обе кривые показывают, что разбавление водяным паром весьма эффективно до его расхода 20% (масс.) на сырье. С утяжелением сырья необходимый расход водяного пара возрастает: если при пиролизе углеводородных газов расход пара равен 10—20%, то при пиролизе бензинов требуется 25—50% пара (в зависимости от фракционного и химического состава бензина). При переходе к керосино-газойлевым фракциям расход пара возрастает примерно до 100%.
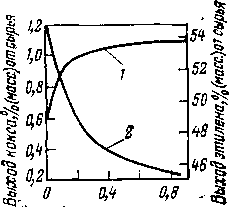
Массовое отношение пар:сырье
Рис. 35. Изменение выхода этилена (1) и кокса (2) при пиролизе пропана в зависимости от разбавления сырья водяным паром.
Данные К. Е. Масальского и В. М. Годика.
На материальный баланс пиролиза благоприятно влияет легкий газообразный разбавитель, поэтому было предложено проводить процесс в присутствии водорода. Он не только является разбавителем — при увеличении концентрации он активно взаимодействует с компонентами реакционной смеси, что объясняется смещением равновесия в сторону соединений, богатых водородом. Достоинство водорода как разбавителя —повышение выхода этилена, снижение выхода ацетилена, тяжелой части смолы и кокса; недостатки процесса — увеличение объема газа и снижение, выхода пропилена.
Промышленное оформление процесса
Основные трудности, связанные с промышленным оформлением пиролиза, такие:
1) необходимость четкого регулирования продолжительности реакции, которая при высоких температурах составляет обычно доли секунды;
2) отложение кокса и сажи в реакционной зоне и при быстром охлаждении газа пиролиза (в «закалочном» аппарате);
3) необходимость применения жароупорных материалов;
4) ограничение пропускной способности установки вследствие значительного удельного объема реакционной смеси, обусловленного высокой температурой, низким давлением и разбавлением сырья водяным паром. Последнее заставляет на установках -средней и высокой пропускной способности иметь несколько (иногда 8—Ю) печей.
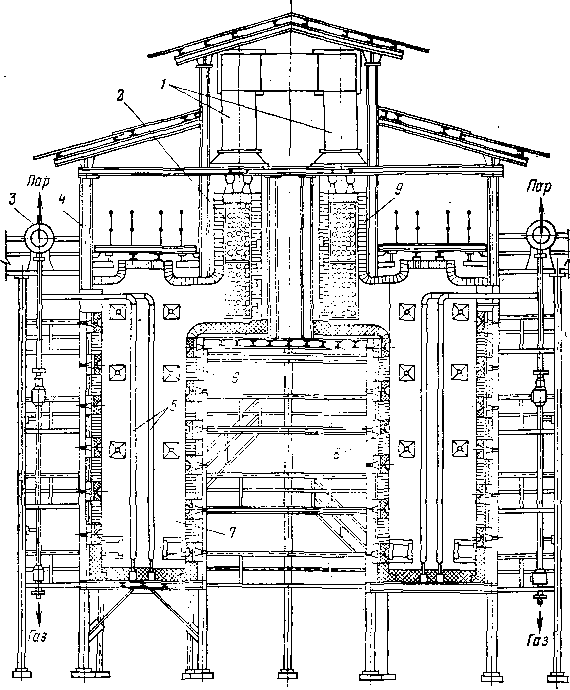
Рис. 36. Печь конструкции ВНИПИнефть для высокотемпературного пиролиза:
/ — газоходы; 2 — конвекционная камера и пароперегреватель; 3 — «закалочно>-испаритель~ ные аппараты и паросборники; 4 — каркас; 5 — реакционный змеевнк; 6 — металлически® кожух и термоизоляция; 7 — радиантная камера; 8 — горелки; 9 — подвесные стены из ша* мотного кирпича.
Совершенствование конструкций трубчатых печей за последние годы позволило снизить время контакта с 2 до 0,25—0,40 с и повысить температуру до 840—870 °С.
Для увеличения относительной поверхности труб (поверхность, приходящаяся на единицу реакционного объема) печи делают многопоточными— с 4—6 параллельными потоками. Объем реакционного змеевика должен обеспечить завершение реакции за короткий промежуток времени. Это достигается интенсивным и ъ то же время -строго равномерным обогревом труб змеевика путем использования печей с панельными горелками и одно- или двухрядным экраном двухстороннего облучения.
Рис. 37. Схема установки пиролиза бензина:
Л/— паровой подогреватель; П-1 — печь; Л-1 — «закалочный» аппарат; Т-2 — котел-утилизатор (закалочно-испарительный агрегат); Е-1 — аппарат масляной промывки; К-1—колонны; Ф-/— фильтры; Х-1 — холодильник: ХК-1— конденсатор-холодильник; С-1 — сепаратор; С-2 — отстойник; К-2 — отпарная колонна.
Трубы в современных печах пиролиза по преимуществу вертикальные, что обеспечивает компактность печи. Печной блок из двух таких печей конструкции ВНИПИнефть, предназначенных для пиролиза бензина и имеющих общую дымовую трубу, изображен на рис. 36. Каждая печь имеет четырехпоточный змеевик диаметром 102 мм; производительность сдвоенной печи 14—16 т сырья в час. Теплонапряженность труб составляет в среднем 222 тыс. кДж на 1 м2 в час, что обеспечивает максимальную температуру стенки змеевика 988 °С и допустимо для труб из стали Х25Н20. Смесь сырья и 50—70% водяного пара проходит часть труб конвекционной секции, размещенной на верху печи. В этой секции сырье должно только -нагреваться (или -нагреваться и испаряться); это соответствует температуре на выходе из секции л;600°С.
Необходимая эффективность процесса достигается, как сказано выше, при -сочетании высокой температуры с малым временем контакта. Применительно к трубчатым реакционным печам это соответствует 840—860 °С и 0,3—0,5 с.
Требуемую глубину превращения сырья можно достигнуть только при высокой теплонапряженности поверхности радиантных труб; эта величина для некоторых печей достигает 335— 378 кДж/(м2-ч) (80—90 тыс. ккал на 1 м2 в час). Интенсивность теплопередачи от внутренней поверхности стенок труб к сырью определяется скоростью сырьевого потока, поэтому многопоточные змеевики пиролизных печей изготовляют из труб уменьшенного диаметра, чтобы увеличить соотношение их поверхности и объема потока сырья. Возможность повышения скорости паров ограничена гидравлическим сопротивлением в трубах и, следовательно, повышением абсолютного давления в зоне реакции, препятствующего процессу. Иногда используют змеевики с переменным сечением, увеличивающимся к выходу, что снижает гидравлическое сопротивление реакционной секции.
На рис. 37 представлена схема установки для пиролиза бензина (схема пиролиза газообразного сырья отличается тем, что водная промывка газов пиролиза заменена масляной и имеется первичная ректификация). Сырье подают насосом при 1 —1,2 МПа в паровой подогреватель Т-1, где оно нагревается до 100 °С; затем сырье смешивают с водяным паром и двумя потоками подают в коллекторы, где поток разветвляется на четыре в каждом коллекторе. Пройдя часть труб конвекционной секции печи П-1, смесь паров бензина и водяного пара поступает в трубы реакционного змеевика. Газ выводят из печи при 840—850 °С и во избежание пиролитического уплотнения непредельных углеводородов подвергают быстрому охлаждению в «закалочном» аппарате А-1. Он представляет собой конденсатор смешения, куда подают водный конденсат. За счег теплоты испарения конденсата температура газа пиролиза снижается до 700 °С. Охлаждение на 140—150 °С достаточно, чтобы за несколько секунд пребывания газа на участке от «закалочного» аппарата до котла-утилизатора Т-2 прекратить реакции пиролиза. Последующее снижение температуры происходит в закалочно-испарительном агрегате (котел-утилизатор), где тепло газов пиролиза используется для производства водяного пара высокого давления.
Газ, охлажденный примерно до 400 °С, проходит еще одну ступень охлаждения в аппарате Е-1, куда подают тяжелое поглотительное масло. Описанная система охлаждения 'входит в печной агрегат, включающий также теплообменник и печь. После аппарата Е-1 все газовые потоки объединяют в общий коллектор, и дальше система отделения смолы от газа общая для всех агрегатов.
Паро-газовую смесь направляют в две параллельно работающие колонны К-1• В нижней части колонн поток отмывают от сажи и кокса тяжелым поглотительным маслом; верх колонн орошается легким поглотительным маслом. В нижней части колонн конденсируется наиболее тяжелая часть смолы, которую откачивают насосом Н-1 через фильтры Ф-1 и после охлаждения в холодильнике Х-1 направляют в аппарат Е-1 и в низ колонн К-1 в качестве тяжелого поглотительного масла, а балансовое количество выводят.
Облегченная паро-газовая смесь с верха колонн К-1 проходит конденсатор-холодильник ХК-1 и, охладившись до 30—35 °С, разделяется в сепараторе С-1 на газ пиролиза, направляемый на компрессию и далее 'на газоразделение, и обводненный конденсат легкого масла, которое отделяют от воды в отстойнике С-2. Легкое масло частично подают на верх колонн К-1, а балансовое количество после отпаривания в колонне К-2 откачивают с установки.
Для переработки тяжелого дистиллятного сырья (вакуумный газойль) требуются более мягкий режим пиролиза и специальные меры, 'направленные на увеличение пробега печи. Так, в практику эксплуатации промышленных уста-новок введена поочередная очистка от кокса одного или нескольких змеевиков печи: из них на несколько часов удаляют сырье и газифицируют образовавшийся кокс водяным паром. При этом достигнута продолжительность пробега 6 мес.
Типичный выход продуктов, полученный пиролизом атмосферного и вакуумного газойлей, в сопоставлении с материальным балансом пиролиза бензина представлен в табл. 10 (стр. 110). Эти данные свидетельствуют о значительном снижении выхода этилена с утяжелением сырья; однако выходы пропилена при низкой жесткости режима сопоставимы, а выходы бутадиена даже несколько выше. Естественно, что с утяжелением сырья выход смолы возрастает.
Глубина превращения сырья при пиролизе и выход целевых продуктов определяются не только температурой и парциальным давлением углеводородных паров на выходе из реакционного змеевика печи, но и профилем температур по длине этого змеевика (при фиксированной температуре на выходе). Профили температур могут иметь вид, изображенный на рис. 38. Выпуклая кривая 1 соответствует быстрому нагреву сырья в начале змеевика до температуры реакции и постоянной температуре при весьма коротком времени, соответствующем максимальному выходу этилена. Однако такой (оптимальный и легко поддающийся оптимизации) ход температур ограничен из-за возможности перегрева труб. При вогнутой кривой 2, т. е. при медленном начальном нагреве, возрастают выходы пропилена и бутиленов за счет некоторого снижения выхода этилена. Кривая 3 иллюстрирует промежуточный случай, соответствующий равномерному нагреву по длине реакционного змеевика.
Для пиролиза помимо печных агрегатов используют реакторы с псевдоожиженным слоем инертного твердого теплоносителя (обычно коксовые гранулы) и с движущимся слоем крупнограну-лированного теплоносителя. Такое аппаратурное оформление предназначено для тяжелого остаточного сырья, которое невозможно пиролизовать в трубчатых печах из-за высокого выхода кокса.
Схема реакторного блока подобна схеме непрерывного коксования с порошкообразным коксом-теплоносителем. Спецификой пиро-
лиза является необходимость сохранить малое время контакта сырья с теплоносителем. Этого можно достигнуть снижением высоты слоя и (или) укрупнением размера частиц теплоносителя, что позволяет увеличить скорость паро-газовой смеси над слоем. Явление перемешивания газовой фазы, свойственное системам с псев-доожиженным слоем, может затруднить регулирование времени контакта, а при завышении этого времени усилятся реакции уплотнения и снизится выход целевых
олефинов. В качестве теплоносителя используют кокс с частицами 0,1—1,5 мм; температура в реакционной зоне 720—770 °С, время реакции 1,2—3,1 с, расход пара 60—260% на сырье.
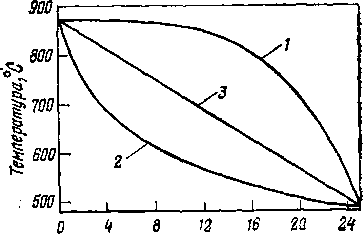
Длина змеевика, м
Рис. 38. Профили температур по длине реакционного змеевика пиролизной печи:
При пиролизе высокосернистого вакуумного гудрона
тол 1 — быстрое нагревание сырья до температурь»
[ПЛОТНОСТЬ lUoU 'КГ/М , СО" реакции; 2 — медленное нагревание;.Л—промежу-
держание серы 5,5% точный случай.
(масс.), коксуемость 14,5% (масс.)] было получено (в % масс.)269:
К концу 70-х годов термоконтактный пиролиз проводили только в масштабе пилотных и единичных опытно-промышленных установок.
Предложен также пиролиз иефтн в потоке газового теплоносителя. Процесс ведут, распыляя нефть в потоке пара, перегретого до 2000 °С. Время пребывания сырья при этой температуре составляет ?»0,02 с. Пиролиз нефти дает примерно 33% (масс.) этилена, 4,4% (масс.) пропилена, 3% (масс.) бутадиена и 14,3%. (масс.) ароматических углеводородов (бензол, толуол, ксилолы); остальное — жидкие продукты (смола) и сухой газ.
Качество и использование жидких продуктов пиролиза
Об использовании газа пиролиза говорилось ранее (стр. 109). Выход жидких продуктов в основном зависит от качества сырья. Ниже приводится средний выход жидких продуктов пиролиза различного сырья (% масс.):
Из этих данных видно, что при пиролизе керосино-газойлевых фракций около половины сырья превращается в жидкие продукты, поэтому только рациональное и полное их использование обеспечит рентабельность установок пиролиза.
Качество жидких продуктов зависит и от сырья и от жесткости режима. Фракционный состав жидких продуктов изменяется с утяжелением исходного сырья. Например, при жестком режиме (860—870°С, 0,3 с, 50% водяного пара) бензиновая фракция 44— 155°С дает 23% (масс.) смолы, содержащей 17,4% углеводородов Сю и выше. В то же время при пиролизе керосино-газойлевой фракции также при жестком режиме (800—840°С, 0,5—0,8 с, 100% водяного пара) образуется 41% жидких продуктов с 60,5% углеводородов Сю и выше270.
После перегонки жидких продуктов с установки уходят четыре фракции: С5 (до 70°С), 70—130°С (бензольно-толуольная), 130— 190°С (С8—Сд) и >190°С (тяжелая смола). Фракция С5 более чем наполовину представлена непредельными; из них примерно 50% приходится на циклопентадиен и изопрен. Циклопентадиен — весьма реакционноспосо&ный углеводород, используемый в ряде синтезов (получение пестицидов, пластификаторов и т.д.). Изопрен — исходное сырье для производства синтетического каучука.
Фракцию 70—130°С подвергают гидрированию (для насыщения непредельных углеводородов), экстракции или адсорбции (для выделения бензола и толуола) и последующему разделению ректификацией. Потребность в бензоле все возрастает, поэтому для увеличения его выхода толуол иногда подвергают дегидроалкилиро-ванию.
Фракция 130—190°С содержит некоторое количество ксилолов и этилбензола (10—12% масс.), «о главными ее компонентами яв-
ляются стирол (до 40%) и другие алкенилароматические углеводороды, а также инден и дициклопентадиен. Благодаря своей высокой химической активности фракция может быть использована для производства полимерных смол, применяемых в шинной и лакокрасочной промышленности. Из тяжелой части смолы на некою- ' рых заводах отгоняют фракцию 190—230 °С с целью выделения нафталина. Тяжелая часть смолы содержит смолисто-асфальтовые компоненты; она используется как сырье для производства сажи или беззольного кокса.
Для жидких продуктов пиролиза характерна высокая концентрация ароматических углеводородов. В легких фракциях присутствуют преимущественно бензол и толуол и в меньших концентрациях углеводороды С8, так как бензол наиболее термически стабилен. Так, во фракции, выкипающей до 190°С (образовавшейся при пиролизе бензина в жестком режиме), обнаружено 33,6% бензола, 22% толуола и 11% ксилолов и этилбензола (в сумме). При этом в более тяжелых фракциях преобладают нафталин и метилнафта-лины, также отличающиеся высокой термической стабильностью.
Другой существенный компонент жидких продуктов — непредельные углеводороды (олефины, циклоолефины и диены). Диены, как известно, весьма нестабильны и способны полимеризоваться и
Таблица 13. Качество жидких продуктов пиролиза
Данные Г. Ф. Лесохиной и соавт.
|
Показатели | Пиролиз беизииа | Пиролиз керосино-газойлевой фракции |
| Плотность р|° |
0,850—0,885 | 0,930—0,960 |
| Йодное число, г 12/100 г Фракционный состав, % (масс.) |
70—85 | 70—86 |
| 12,0 | ||
| и. к — 70 °С | 5,0 | |
| 70— 130 °С | 50,0 | 21,5 |
| 130—190 °С | 19,1 |
17,5 |
| 190—250 °С |
5,5 | 18,2 |
|
>250°С | 13,4 | 37,8 |
| >350°С | 4,3 | — |
| >450 °С | — ¦¦ | 12,8 |
| Элементный состав, % (масс.) | ||
| С |
89—90 | 90,8 |
|
Н | 10—П | 8,7 |
| S | 0,1 | 0,56—0,60 |
|
Содержание ароматических углеводородов*, % (масс.) на смолу | 27,5 (6,3) | |
| бензол | 10,9 (4,5) | |
|
толуол | 18,3 (4,2) | 7,9 (3,2) |
| ксилолы и этилбеизол | 9,2 (2,1) |
3,8 (1,6) |
| Выход жидких продуктов, % (масс.) иа сырье |
23,0 | 41,0 |
|
пиролиза |
* В скобках приведены данные в % (масс.) на исходное сырье.
окисляться на воздухе. Содержание остальных углеводородов 'Невелико, что подтверждается и 100%-ной сульфируемостью жидких продуктов.
В табл. 13 приведены некоторые свойства жидких продуктов, получаемых пиролизом бензина (смесь 75% прямогонного и 25% рафината) и керосино-газойлевой фракции.
РЕКОМЕНДУЕМАЯ ЛИТЕРАТУРА
Магарил Р. 3. Механизм и кинетика гомогенных термических превращений углеводородов. М., Химия, 1970. 224 с. ,
Товарные нефтепродукты. Свойства и применение. Справочник/Под ред. В. М. Школьникова. М., Химия, 1978. 472 с.
Сюняев 3. И. Производство, облагораживание и применение нефтяного кокса. М., Химия, 1973. 296 с.
Гюлъмисарян Т. Г., Гилязетдинов Л. П. Сырье для производства углеродных печных саж. М., Химия, 1965. 160 с.
Паушкин Я. М., Адельсон С. В., Вишнякова Т. П. Технология нефтехимического синтеза. М., Химия, 1973. Ч. I, 448 с.
Лесохина Г. Ф., Мухина Т. Н., Ходаковская В. А. Состав и переработка жидких продуктов пиролиза на отечественных установках. М., ЦНИИТЭнефте-хим, 1977. 88 с.
РАЗДЕЛ ТРЕТИИ
ТЕРМОКАТАЛИТИЧЕСКИЕ ПРОЦЕССЫ